Bronchiectasis: a new dawn in diagnosis and treatment
Lung diseases
Breathing problems
Diagnosis of bronchiectasis has increased with broader availability of high-resolution CT scanning. Early diagnosis and development of a patient-specific exercise and airway clearance regimen and a pathogen eradication and suppression plan are key to successful treatment. Management can be shared by GPs in co-operation with a specialist respiratory physician and respiratory physiotherapist.
- Underdiagnosis of bronchiectasis has been a significant problem caused in part by clouding by coexisting respiratory comorbidities including chronic obstructive pulmonary disease and asthma.
- High-resolution CT scanning is now widely available and is the gold standard diagnostic test.
- Confirming a diagnosis of bronchiectasis enables the provision of disease-appropriate treatments and the rationalisation of potentially inappropriate treatments.
- Exercise, airway clearance and mucoactive therapies all help reduce airway inflammatory burden, leading to reduced symptoms and exacerbations and improved quality of life.
- Microbiological surveillance enables the identification of agents such as Pseudomonas aeruginosa and nontuberculous mycobacteria that have powerful negative impacts on symptoms, lung function, exacerbations and healthcare utilisation.
- Antibiotics play a key role in the eradication of novel microbial isolates, immunomodulation, exacerbation management and long-term suppression of microbial colonisation in the airways.
- GPs can share in tailoring a patient-specific management plan in co-operation with a specialist respiratory physician and respiratory physiotherapist.
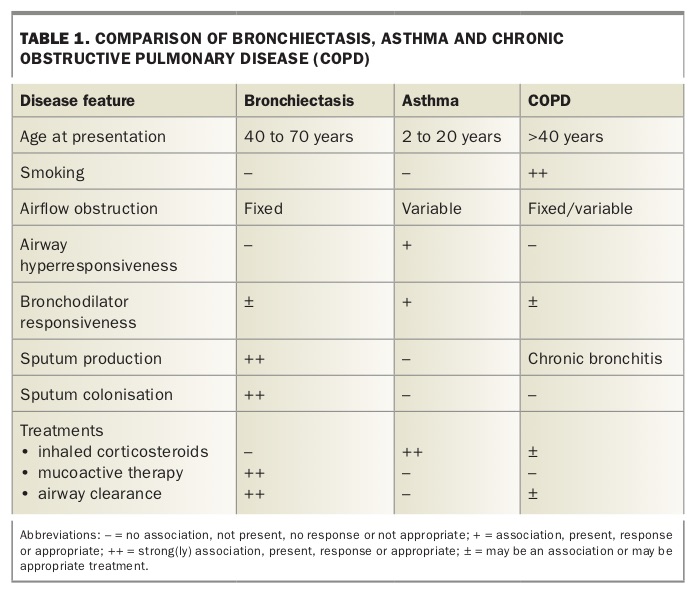
Bronchiectasis shares many symptoms in common with other respiratory conditions including chronic obstructive pulmonary disease (COPD), bronchitis and asthma (Table 1) and the risk of underdiagnosis may persist if the diagnosis is not considered and radiologically confirmed. Bronchiectasis can be defined simply as a permanent dilatation of the airways, arising from chronic bronchial inflammation and infection.1 Before high-resolution CT scanning became broadly available, infection may have been apparent to a diagnosing clinician but pathological bronchial dilatation was difficult to confirm, meaning that this condition remained largely underdiagnosed.
{kind=link}
When misdiagnosis persists, patients with bronchiectasis tend to be regarded as having ‘difficult’ or treatment-resistant asthma or COPD. As such, they are often, sometimes mistakenly, treated with increased doses of inhaled corticosteroids and bronchodilators, and do not receive the physiotherapy, mucoactive therapy and antibiotics that are appropriate for treating bronchiectasis.
Historically, non-cystic fibrosis (non-CF) bronchiectasis has attracted few advocates from the medical, pharmaceutical or broader community and has therefore been considered an orphan disease. More recent initiatives have substantially increased interest and research in non-CF bronchiectasis, leading to a strengthening evidence base for therapies and the development of a national bronchiectasis registry (https://lungfoundation.com.au/research/our-research/bronchiectasis). Guidance for health professionals who are providing care to children or adults with bronchiectasis is available in the form of Australian and international guidelines and other resources derived from them (Box 1).2,3 Seeking and confirming a diagnosis of bronchiectasis allows the provision of disease-appropriate treatment, with major potential benefit in both disease prognosis and quality of life for those with this condition.
{kind=link}
Incidence, morbidity and mortality
Globally, there has been a substantial increase in the recognition and diagnosis of bronchiectasis. Prevalence data for bronchiectasis in Australia are scant, yet this condition may affect up to 1.5% of children aged 15 years and under who are referred to respiratory physicians. Population estimates vary from four to 1470 for every 100,000 children in Australia and New Zealand.4
In 2015, bronchiectasis was diagnosed in more than 17,000 hospitalisations in Australia and hospitalisation rates for females were double male rates. Hospital length of stay for bronchiectasis is typically double the duration of all other hospitalisations in Australia and has significant associated increases in cost estimates.5 Bronchiectasis as a primary or co-diagnosis tends to prolong hospital length of stay, increases the likelihood of admission to the intensive care unit and mortality in those with coexistent COPD and is associated with an increased risk of a first cardiovascular event.6,7 Review of observational studies has suggested that bronchiectasis may coexist with and complicate COPD (in 54% of cases) and uncontrolled severe asthma (in 34%).8,9
CF has an incidence of one in 2000 live births in Australia, and children with the condition are usually affected by bronchiectasis. A family history of bronchiectasis is therefore important, as CF, primary ciliary dyskinesia and Kartagener’s syndrome share an autosomal recessive inheritance pattern.
Globally, Indigenous populations, and particularly Indigenous Australian children, are at significantly increased risk of bronchiectasis and chronic suppurative lung disease and share a disproportionately large burden of disease and number of associated hospital admissions.10,11 Importantly for these children, appropriate early management may make chronic suppurative lung disease largely preventable. A further important risk group is patients with established lung disease; bronchiectasis has been identified in up to 50% of patients with COPD.12,13
Quality of life is substantially impaired in patients with bronchiectasis and is further adversely affected during exacerbations. Mortality among bronchiectasis cohorts has been identified as being as high as 16 to 30% over a decade, mainly affecting the elderly.14
Pathology
Pathologically, bronchiectasis is a heterogeneous condition in which follicular, saccular and atelectatic histological patterns have been described.15 This pathological diversity may suggest that pathological bronchiectatic airway dilatation may be the final common pathway of several infective and immunological airway insults. The follicular pattern appears most frequently, usually involving the smaller airways. In the mildest forms, subepithelial lymphoid aggregations and oedema are the only findings.
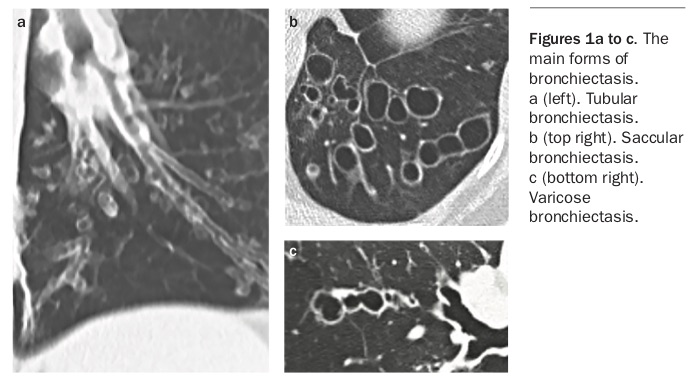
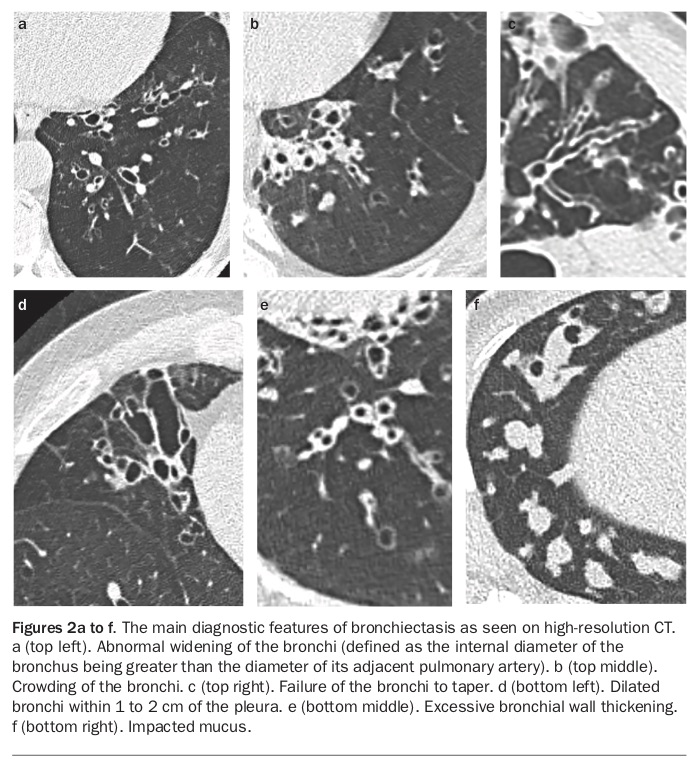
In more severe disease, there is evidence of inflammatory cell aggregation, with macrophages, lymphocytes and monocytes within the airway walls and neutrophils predominating in the airway lumen. More severe disease is associated with progressive destruction and loss of elastin, smooth muscle, mucous glands and cartilage. Lymphoid follicles attain such size that in places they lead to regional obstruction of small airways with distal collapse. The essential lesions are destructive mural bronchitis, bronchiolitis and interstitial inflammation.15Airflow obstruction affecting both large and small airways is a key feature of bronchiectasis and results from loss of structural integrity as well as inflammatory infiltrate within the airway wall. This airflow obstruction inhibits the clearance of sputum, bacteria and inflammatory mediators expressed in response to airway microbiota. The gross pathology of the various forms of bronchiectasis are illustrated in Figures 1a to c, and the diagnostic features in Figures 2a to f.
{kind=link}
{kind=link}
Infection and inflammation – the vicious cycle
Host defence in the lung is maintained by both innate and adaptive immune mechanisms and a major contributor is the maintenance of an effective mucociliary barrier. This barrier is comprised of airway mucus that traps inhaled foreign matter, including microbiota, and a ciliary escalator that drives mucus and entrapped material upwards for subsequent clearance by expectoration. These protective mechanisms are broken down in conditions such as chronic bronchitis, emphysema and bronchiectasis, which variably cause pathological distortion and destruction of the normal airway anatomy, mucus hypersecretion and subsequent disruption of mucociliary clearance from the lung.
In the acute setting, bacteria in the airway or distal alveoli may cause lower respiratory tract infections. In the structurally normal lung, resolution of infection is associated with clearance of the airway of all the involved bacteria, inflammatory cells and mediators. In the structurally damaged lung, however, these clearance mechanisms fail, and airway colonisation, infection and inflammation persist.
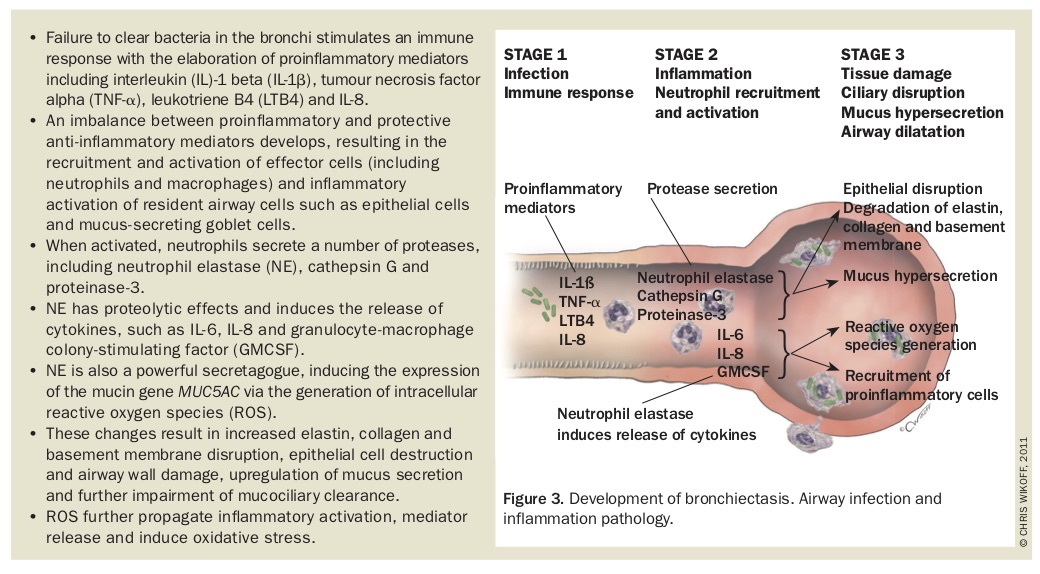
Dysfunction of the respiratory epithelium (ciliary dyskinesia) and depletion or dysfunction of immune mediator cells and molecules, including immunoglobulin (Ig) G and IgE, complement (due to mannose- binding lectin deficiency), cathelicidins (antimicrobial peptides), toll-like receptors (which are key to pathogen recognition), neutrophils, macrophages, and T and B lymphocytes, may permit microbial persistence, excessive inflammatory response to infection and failure of mucociliary clearance. It is not surprising, therefore, that bronchiectasis may result from several pathological insults that are disparate but unified by their impact on depletion of immune function, promotion of airway inflammation and impairment of mucociliary clearance. The diseases frequently associated with bronchiectasis are listed in Box 2. The process of development of bronchiectasis is expanded in Figure 3.
{kind=link}
{kind=link}
If this cycle of infection and inflammation is uninterrupted then progressive lung injury occurs, leading to worsened lung function and ongoing inflammation promoting both symptoms and pulmonary exacerbation. The key to breaking this cycle therefore resides in clearance of proinflammatory bacteria and the downstream inflammatory mediators from the airways to reduce the overall inflammatory burden and consequent lung damage.
Symptoms and presentation
The diagnosis of bronchiectasis should be considered in children and adults with cough and chest symptoms suggestive of chest infection, particularly when there is a history of purulent sputum production, a family history of bronchiectasis or a history of pneumonia in infancy or childhood. There are no pathognomonic features in bronchiectasis so a high index of suspicion is required.
The incidence of bronchiectasis is higher among females and the elderly, and symptoms suggestive of bronchiectasis should be investigated in these patient populations in particular. Features suggesting bronchiectasis can include persistent or recurrent productive cough; recurrent chest infections; infections that are slow to resolve and persistent chest x-ray abnormalities despite antibiotic treatment. The coexistence of COPD, asthma or other respiratory disease with prominent cough and sputum production should also raise the suspicion of bronchiectasis.
Other suggestive features include haemoptysis (often streaking), cough-induced chest pain, poor nutritional status, failure of response to inhaled corticosteroids or bronchodilators in suspected asthma/ bronchitis, and recurrent sputum isolates of Haemophilus or Staphylococcus species or a single sputum isolate of Pseudomonas species or methicillin-resistant S. aureus. Clinical signs that may be found include finger clubbing, halitosis (commonly associated with anaerobic infection), hyperinflation of the chest and poor nutritional status.
Causation and association – diagnostic investigation
The initial challenge in the diagnosis of bronchiectasis is considering it as an explanation for the presenting symptoms. The diagnosis itself is achieved relatively simply on high-resolution CT (HRCT) scanning of the chest; the subsequent challenge is determining the cause and severity.
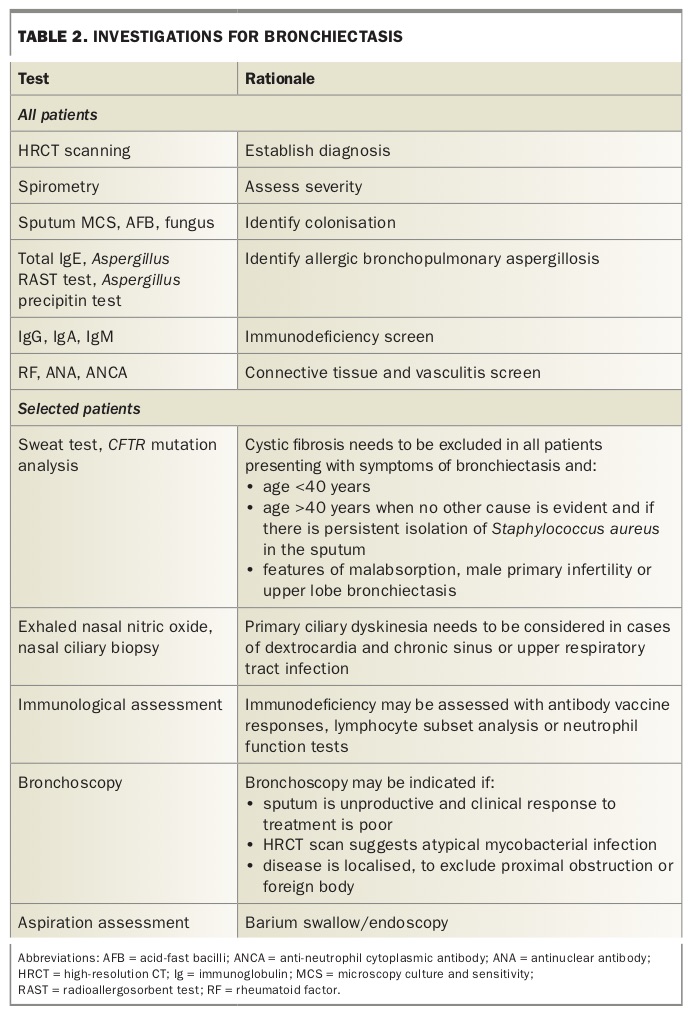
Diagnostic studies include serological, biochemical, microbiological, radiological and physiological investigations (Table 2). Several aetiological factors contribute in the development of bronchiectasis (Box 2). Identification of these factors has been shown to be important for directing management.16 This is perhaps most evident in immunodeficiency syndromes including antibody deficiency, such as common variable immunodeficiency and X-linked agammaglobulinaemia, for which intravenous immunoglobulin replacement therapy may revolutionise outcomes.
{kind=link}
Testing for cystic fibrosis and primary ciliary dyskinesia in adults under 50 years of age is beneficial.1 The presence of gastro-oesophageal reflux may be identified using barium or gastrografin swallow imaging. Studies of children and adults show that the identification of a cause leads to a change in management (to disease-specific management strategies) in more than half of patients.
Identifying a cause remains difficult in patients with non-CF bronchiectasis. Even after careful evaluation up to 40% of cases remain classified as idiopathic.
Radiology
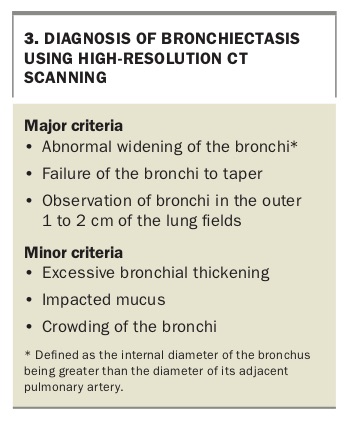
HRCT scanning is the radiological diagnostic gold standard test for bronchiectasis and increased availability has increased confirmation of this diagnosis. The main findings on HRCT (Box 3) that indicate bronchiectasis are abnormal widening of the bronchi (defined as the internal diameter of the bronchus being greater than the diameter of its adjacent pulmonary artery), failure of the bronchi to taper and observation of bronchi in the outer 1 to 2 cm of the lung fields (Figures 2a, 2c and 2d). Multidetector HRCT has elevated diagnostic yield with shorter acquisition times and reduced motion artefact. CT scanning without high-resolution acquisition can either miss or significantly underestimate the presence and extent of bronchiectasis.
{kind=link}
The distribution of lung involvement can also be determined by CT and may be suggestive of aetiology. CF (bilateral) and tuberculosis (often unilateral and upper lobe) typically cause upper lobe involvement; allergic bronchopulmonary aspergillosis causes more proximal bronchial involvement. Middle lobe involvement has been observed in the setting of lymph node enlargement with bronchial obstruction, and is also associated with atypical mycobacterial infection with possible additional radiological findings of peripheral infiltrate and mucus plugging. Lower lobe involvement appears to be a feature in aspiration, ciliary dyskinesia, hypogammaglobulinaemia and childhood respiratory infection, although these findings have not been universal.
Lung function
Spirometry, including measurement of forced expiratory volume in one second (FEV1), forced vital capacity and bronchodilator responsiveness, should be performed in all patients at diagnosis and repeated in monitoring on an annual basis. A significant increase (greater than 12%) in FEV1 or forced vital capacity after bronchodilator use may be seen in up to 30% of patients tested and may indicate asthma or clinically significant reversible airflow obstruction. These patients should be treated with bronchodilators and, if necessary, low-dose inhaled corticosteroids.
Microbiology
Sputum culture should be performed at diagnosis to ascertain colonisation and to direct subsequent antibiotic therapy. Nontypeable H. influenzae, Streptococcus pneumoniae and Moraxella catarrhalis are the most common isolates identified in children and adults (in about 35% of patients), followed by P. aeruginosa (in about 5 to 30% of patients). Isolates of mycobacteria and Aspergillus species are found in few patients, but when found may adversely impact prognosis and will require specific treatment. The geographical prevalence of specific microbial isolates varies markedly. Studies in Spain and the US have suggested nontuberculous mycobacterial prevalence of between 8.3 and 63%.17,18
Changes in microbial flora over time are a feature of bronchiectasis and surveillance with annual follow up is necessary. Pseudomonas acquisition is a feature of late bronchiectasis and once colonisation has occurred eradication may prove difficult. Sputum cultures are usually adequate for testing but pharyngeal swabs may occasionally be needed in children. Bronchoscopy to obtain specimens for microbiological culture is only rarely required.
P. aeruginosa is a highly evolved Gram-negative bacteria characterised by its use of biofilms and the development of complex antimicrobial resistance mechanisms, making treatment challenging.19 Acquisition of P. aeruginosa is a significant event and is associated with about a threefold increased risk of death, sixfold increase in hospital admissions, doubled rate of exacerbations and poorer quality of life. Additionally, healthcare costs have been shown to increase by 87% in the 12 months after colonisation.20,21 This risk highlights the importance of the active management of patients to minimise cross infection within clinical consulting spaces.22
Standard microbial cultures are selective and identify a limited range of bacterial species in clinical samples. Newer, culture-independent techniques employ molecular approaches such as 16S rRNA gene sequencing to describe the airway microbiome and have been shown to detect a much greater variety of microbes than standard culture techniques.23 Such techniques may become increasingly important as predictors of outcome (and possibly exacerbations) in the airway since it has been recognised that perturbations of microbial interactions within the microbial community of the lung microbiome appear to be affected by disease severity, airway obstruction and exposure to antibiotics.
Severity measures
The development of a bronchiectasis severity score has been addressed in several studies with the purpose of modelling prognosis. The FACED score was designed to predict five-year mortality using five key variables:
• F: FEV1, score 0 to 2
• A: Age, score 0 to 2
• C: Colonisation with P. aeruginosa, score 0 to 1
• E: Extent of radiological involvement (number of lobes affected), score 0 to 1
• D: Dyspnoea (Medical Research Council dyspnoea scale), score 0 to 1.24
Three severity groups, mild (0 to 2 points), moderate (3 to 4 points) and severe (5 to 7 points), predicted five-year respiratory mortality of 0.9 to 2.4%, 13.6 to 15.3% and 45.6 to 51.6%, respectively. This is equivalent to five-year respiratory mortality rates of about one in 50 patients with mild disease, one in six patients with moderate disease and one in two patients with severe disease.
In this model, neither exacerbations (hospital admissions) nor nontuberculous mycobacterial colonisation were predictive of mortality, and age and FEV1 were the strongest mortality predictors. The FACED score and the similar, although slightly more detailed, Bronchiectasis Severity Index are being further evaluated for their ability to predict clinically important disease-related outcomes, including exacerbation frequency, quality of life, respiratory symptoms, functional capacity and lung function decline in bronchiectasis.25,26
Specialist management
Some patients with bronchiectasis will benefit from ongoing management by a respiratory physician. Specialist units may develop management plans at the time of diagnosis for both maintenance care and exacerbations. These plans help identify clear treatment goals and measures. Patient groups and conditions for which ongoing specialist management should be considered include:
• all children with bronchiectasis
• chronic P. aeruginosa, opportunist mycobacteria or methicillin-resistant S. aureus colonisation
• poor and declining lung function
• recurrent exacerbations (more than three per year)
• patients receiving or in need of prophylactic antibiotic therapy (oral or nebulised)
• complex associated comorbidity such as allergic bronchopulmonary aspergillosis, rheumatoid arthritis, immune deficiency, inflammatory bowel disease and primary ciliary dyskinesia
• patients in whom lung transplantation is being considered.
Treatment
The objectives of good management in bronchiectasis are to:
• improve wellbeing
• reduce symptoms and exacerbations
• improve quality of life
• maintain or defer deterioration in lung function
• prolong survival.
Developing an appropriate management plan requires application of robust primary care principles as well as engagement of a specialist multidisciplinary team experienced in the management of non-CF bronchiectasis (Box 4).
{kind=link}
All patients diagnosed with challenging bronchiectasis should be referred to a respiratory physician and a physiotherapist experienced in airway clearance techniques for assessment, which may assist in confirming the diagnosis, disease severity, comorbidities and complications and in further developing management and self-management plans. Management plans need to be individualised, tailored to patient preference and reviewed on an annual basis.
Inhaled corticosteroids and beta agonists
In common with asthma, a significant proportion of patients with bronchiectasis have bronchodilator reversibility (15 to 36%) and evidence of ongoing inflammation.27,28 Asthma remains common and bronchodilators should be prescribed for empirical trial in true cases of asthma. Bronchodilators can also be used effectively before mucoactive therapy and airway clearance, although in the absence of bronchodilator reversibility and subjective benefit there is little evidence of benefit in routine use.
Inhaled corticosteroids are frequently used in patients with bronchiectasis yet their benefit is far from certain. Meta-analysis suggests they have no significant beneficial effect on lung function, exacerbation frequency or quality of life and therefore should not be prescribed for bronchiectasis.29 Furthermore, inhaled corticosteroids in those with chronic respiratory conditions can increase the risk of nontuberculous mycobacterium infections, highlighting the care needed when deciding to prescribe them for patients with bronchiectasis.30 Inhaled corticosteroids may be indicated in the reasonably large proportion of patients with bronchiectasis who have concomitant airways disease (COPD or asthma). In the absence of these co-diagnoses, active efforts should be made to withdraw or titrate inhaled corticosteroid use if already commenced.
Mucoactive therapy
Mucoactive agents provide mucolysis, mucokinesis and augmented expectoration. Mucoactive therapies are aimed at improving mucus quality by changing the physiochemical properties of mucus, reducing tenacity and improving flow characteristics, thereby improving mucociliary clearance. Some agents are listed in Table 3.
{kind=link}
Mucoactive therapies have been largely proven to be effective in treating CF but there is little evidence for their effectiveness in non-CF bronchiectasis. Important differences exist between CF and non-CF bronchiectasis. These are perhaps best exemplified by the mucolytic agent dornase alfa (recombinant human DNase), which is widely used and effective in CF but has been shown to be ineffective and possibly detrimental in non-CF bronchiectasis. Evidence of a drug’s efficacy in non-CF bronchiectasis is therefore needed, rather than making assumptions about its efficacy based on the CF literature and experience.
Nebulised short-acting beta-2-agonist bronchodilators have been shown to improve sputum volume production and may be a useful adjunct to mucoactive treatment. Both inhaled mannitol and hypertonic saline may cause bronchoconstriction, so a monitored test dose is required at first use, with measurement of FEV1 before and after nebulisation. Mucoactive therapy regimens usually need to be implemented, reviewed and revised after two to three months. The expertise of experienced physiotherapists is invaluable in this regard.
Physiotherapy
Traditional approaches to physiotherapy in bronchiectasis relied largely on postural drainage and chest percussion. Both of these strategies are now outdated in adults as they inhibit autonomous therapy and potentially exacerbate position-dependent gastro-oesophageal reflux. Chest percussion retains some benefit in paediatric management.
Positive expiratory pressure (PEP) strategies enhance and promote mucus clearance by preventing airway collapse through stenting the airways and increasing intrathoracic pressure distal to retained secretions. The simplest PEP therapy is bubble PEP, in which a patient exhales through a hose into a bottle containing 10 to 12 cm of water. An oscillating pressure is created by the movement of the column of water. Commercial PEP and oscillating PEP devices comprise a mask or mouthpiece with a one-way valve connected to either a small-exit orifice (PEP) or, more commonly, an adjustable expiratory resistor (oscillating PEP). Information about these devices is available online at http://bronchiectasis.com.au/physiotherapy/ techniques/oscillating-positive-expiratory-pressure-therapy. PEP therapy is generally performed once or twice daily, usually in combination with mucoactive treatments.
Airway clearance programs should be instituted in all patients with bronchiectasis who have daily sputum production and should be especially reinforced during periods of exacerbation. Occasionally, exercise can substitute for airway clearance; the likely contribution of a particular form of exercise to airway clearance is best assessed by a trained physiotherapist.
Antibiotics
Antibiotics are central to the management of suppurative lung disease and are used in three different indications:
• to attempt eradication of new airway isolates
• to treat acute infection or exacerbation
• as a long-term maintenance for suppression of chronic colonisation.
Eradication of airway colonists
As mentioned previously, the microbial flora of the airway changes over time in patients with bronchiectasis. Strategies to eradicate new airway colonists appear useful in patients with CF bronchiectasis but evidence of benefit in those with non-CF bronchiectasis is still awaited.
Antibiotic strategies for the long-term suppression of microbial colonisation are now more flexible and may include intermittent or continuous use of a single oral agent, intermittent or continuous use of alternating oral agents and/or intermittent or continuous use of nebulised antibiotics. However, key to the development of an antibiotic strategy is the identification and nature of the airway colonisation. These strategies can be complex and protracted and are best developed by specialist respiratory units.
Exacerbations
Antibiotic treatment of acute infection in the structurally damaged lung may need to be protracted. Home-treated patients may in some cases need antibiotic courses extended to one to two weeks, and hospital-treated patients may need seven to 14 days of intravenous antibiotic treatment. It is important to identify Pseudomonas colonisation, (present in about 30% of patients with bronchiectasis) because this infection will be largely unresponsive to beta-lactam monotherapy and may represent a risk for further deterioration in lung function. The use of long-term oral antibiotic treatment is inappropriate for most patients with bronchiectasis. It may be required in those suffering recurrent exacerbation, although only after confirming the adequacy of and adherence to airway clearance and mucoactive treatments.
Exacerbations are a recurrent feature of bronchiectasis and have substantial detrimental impacts on symptoms, quality of life, treatment cost and decline in lung function. Exacerbation is suggested by increase in frequency of cough, change in cough character, fever and increase in sputum volume and purulence. Dyspnoea and exertional limitation may be a feature, along with malaise, lethargy and anorexia. Although many patients have worsening chest auscultatory findings during exacerbation, spirometry is not usually acutely depressed.
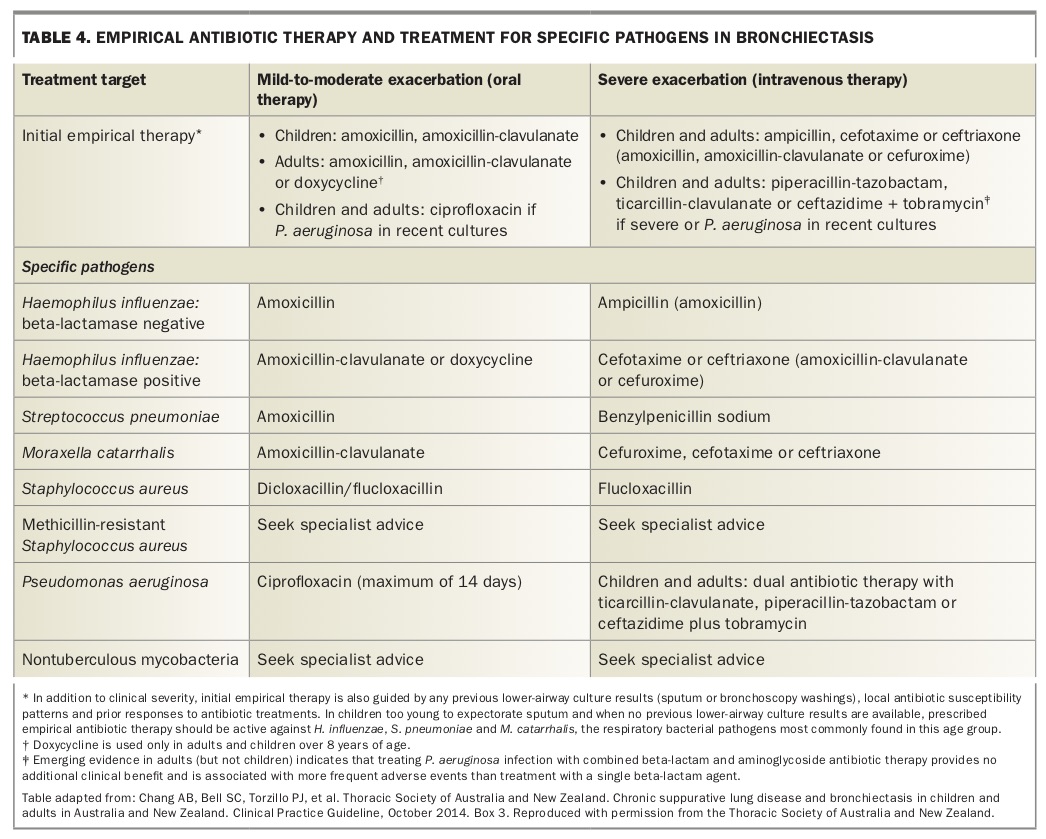
Sputum culture should be performed before antibiotic therapy is started; empirical treatment should be commenced rather than waiting for the result. Treatment should be directed at the last known isolate while recalling the efficacy of the last treatment provided. If no previous microbiological guidance is available, an empirical approach is warranted (Table 4).
{kind=link}
Antibiotic selection can be challenging when multiple pathogens are present, although quantitative assessment of a culture can be directive. New pathogens generally require treatment although a period of observation may be warranted if no clinical sequelae accompany a novel acquisition. Treatment needs to be commenced promptly at the onset of an exacerbation; educating patients about recognising an exacerbation and providing a prescription to enable self-initiation of treatment can facilitate this.
Although exacerbations of lower respiratory tract infections are common, so too are infections such as atypical pneumonia. Minimal chest x-ray evidence may be found in atypical pneumonia, and symptoms such as headache, myalgia, scant sputum production and deranged liver function test results should prompt consideration of macrolide therapy for mycoplasma or chlamydia infection. Similarly, common comorbidities such as sinusitis, rhinitis, asthma and gastro-oesophageal reflux disease may exacerbate symptoms and may need independent treatment at the time of exacerbation.
Referral for intravenous therapy and inpatient treatment may be needed if there is high fever, significant constitutional symptoms such as malaise, anorexia and weight loss, dehydration, difficulty with sputum clearance or decline in lung function. A limited capacity to initiate, continue or escalate a program of mucoactive therapy and airway clearance also indicates that referral is appropriate.
Inhaled antibiotics offer an exciting treatment opportunity in non-CF bronchiectasis for the suppression of bacterial colonisation, treatment of acute exacerbation and potentially in the eradication of colonist pathogens. There has been much recent trial activity exploring the use of antibiotics including ciprofloxacin, tobramycin, colistimethate sodium, aztreonam, amikacin and ceftazidime, and although the results are not universally positive they suggest some capacity to reduce bacterial burden, reduce exacerbation frequency and facilitate pathogen eradication.31 Inhaled antibiotics can achieve high concentrations at the site of infection while minimising systemic side effects and showing good patient tolerability characteristics. To date there is little evidence to suggest the emergence of treatment-resistant pathogens with their use.32 Vibrating mesh technologies in newer nebulisers are faster than conventional jet or ultrasonic nebulisers and provide shorter treatment times and more efficient pulmonary deposition of drug particles.
Macrolide antibiotics
Macrolide antibiotics have proven immunomodulatory benefits in patients with bronchiolitis and CF bronchiectasis and evidence of benefit is emerging in non-CF bronchiectasis. Meta-analysis of macrolide impact in bronchiectasis has shown clear protection against exacerbation (odds ratio, 0.31; 95% CI, 0.08 to 1.15) and improvement in quality of life.33 Macrolides are clearly effective in reducing exacerbation frequency but it is not yet clear which macrolide regimen is most effective, as head-to-head comparisons and meta-analyses have not been done. A systematic review suggested that azithromycin may be more effective than erythromycin or roxithromycin, although it was associated with diarrhoea and abdominal pain, which can be limiting in some patients.34
Azithromycin is an important agent in the management of nontuberculous mycobacteria and as such should be avoided in those with previous nontuberculous mycobacteria isolates to avoid resistance concerns. A pretreatment ECG is recommended as azithromycin can be associated with proarrhythmogenic QT prolongation.
Exercise and rehabilitation
Reduced exercise capacity in bronchiectasis is likely to be due to sputum production, progressive airflow obstruction, chest hyperinflation and dyspnoea, altered respiratory mechanics, decreased skeletal muscle bulk and ongoing infection. Bronchodilator therapy and control of infection improve exercise capacity and functional level but these may be further augmented by exercise, such as that included in pulmonary rehabilitation programs. Exercise such as walking, cycling and trampolining can also lead to full lung inflation and contribute to effective expectoration and mucus clearance. Improved exercise capability appears to be linked with improved quality of life in patients with non-CF bronchiectasis and should be encouraged.
Vaccination
Vaccination against pathogens known to trigger respiratory exacerbation and decrease respiratory tract infections has become standard practice in the care of children and adults with chronic lung disease, but there is little evidence to support this in patients with non-CF bronchiectasis. Nevertheless, clinical experience would recommend that both influenza and pneumococcal vaccinations be made available to all patients with established bronchiectasis.
Conclusion
Bronchiectasis causes substantial morbidity, mortality and impairment in quality of life. The broader availability of high-quality HRCT scanning has seen a significant increase in diagnostic capture. The key to successful treatment lies in early diagnosis, a patient specific exercise and airway clearance regimen and the provision of a management plan for pathogen eradication and suppression with flexible responsiveness for acute exacerbations. Challenges for the future include better definition of disease mechanisms and the confirmation of evidence defining and improving of antibiotic, mucoactive and airway clearance strategies. MT