Persistent pruritic purple papules
Test your diagnostic skills in our regular dermatology quiz. What are these itchy lesions, which first began to appear on a patient’s back and hands?
Case presentation
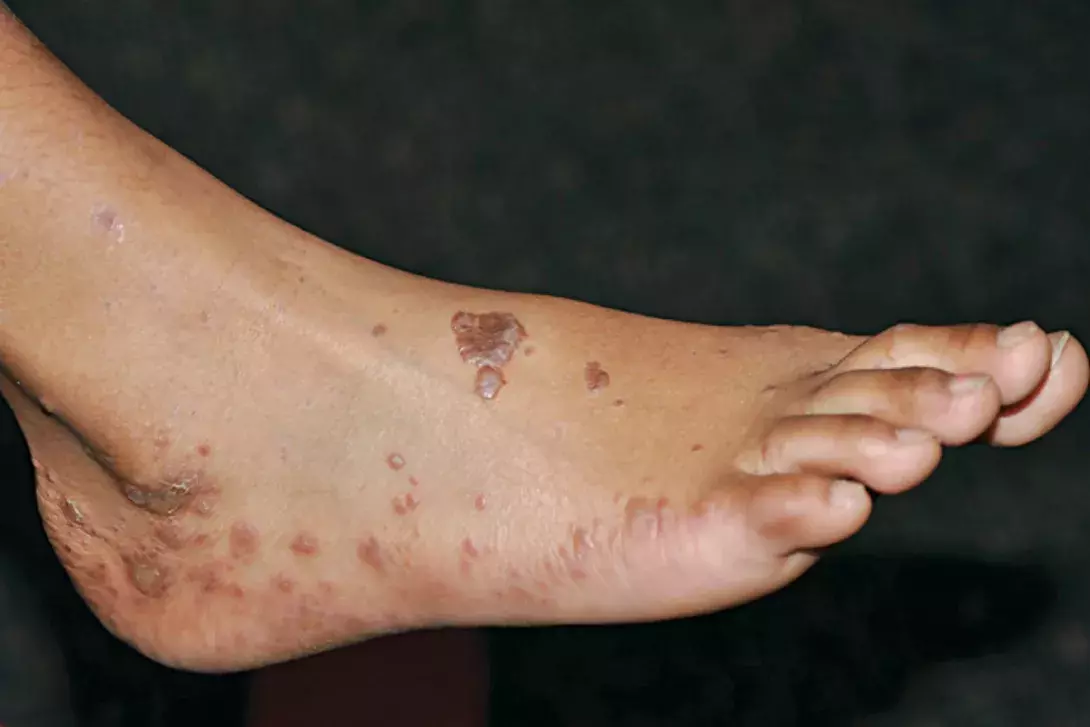
A 48-year-old woman presents with a two-month history of intensely itchy papules on her back and hands. More recently, the lesions have begun to appear on her lower limbs and feet (Figure), occurring over bony prominences and after minor trauma. The lesions are hyperpigmented, violaceous, polygonal papules with a flat top.
{kind=link}
Differential diagnoses
Conditions to consider among the differential diagnoses include the following.
- Granuloma annulare (GA). This benign, self-limiting, inflammatory dermatosis of unknown aetiology has a predominance in females and more commonly affects those under the age of 30 years.1 The lesions are almost always asymptomatic and most commonly involve the upper and lower limbs, particularly the hands, elbows and knees.1 The most common form is localised GA, which presents with small groups of nonpruritic flesh-coloured dermal papules that spread slowly to form annular lesions; generalised GA is characterised by hundreds of disseminated, small, skin-coloured papules that spare the scalp and acral surfaces.1 Perforating GA lesions are skin-coloured papules with central umbilication that preferentially involve the upper extremities, whereas subcutaneous GA lesions are larger flesh-coloured dermal nodules that can involve acral surfaces.1 Biopsy is diagnostic and shows necrobiotic collagen degeneration with surrounding palisading mixed inflammatory infiltrate consisting of monocytes, histiocytes, lymphocytes, fibroblasts and giant cells.1
- Sarcoidosis. Cutaneous sarcoidosis may be confined to the skin or be the first sign of systemic disease. Sarcoidosis is an idiopathic, noncaseating-granulomatous, multisystem disease. About 30% of patients with cutaneous sarcoid develop systemic sarcoidosis within two years.2 The most common clinical manifestation of cutaneous sarcoidosis is the maculopapular form, typified by red, brown and violaceous macules/papules that involve the face (periorbital area, eyelids, nasolabial folds). They may be associated with chest radiographic changes, which occur in stage I sarcoidosis (granulomas in lymph nodes only) and II sarcoidosis (granulomas in lymph nodes and lung).2 Cutaneous sarcoidosis may involve old scars or areas of trauma and this often correlates with degree of visceral sarcoid disease activity. Lupus pernio is a worrisome harbinger of chronic fibrotic pulmonary disease and presents with red to purple indurated lesions affecting the face, ears and nose, with extension into the nasal mucosa sometimes causing septal perforation.2 Other clinical forms of cutaneous sarcoidosis include pruritic papules in photoexposed areas, scarring alopecia, and lichenoid eruptions, although these are less common.2 Lofgren’s syndrome is a form of sarcoidosis in which there is the triad of erythema nodosum, hilar lymphadenopathy and arthritis or arthralgia. The histopathological features of sarcoidosis, which are diagnostic, include dermal, noncaseating epitheloid histiocytes with surrounding lymphocytic and monocytic infiltrate, also known as sarcoidal noncaseating granulomas.2
- Prurigo nodularis (PN). This chronic idiopathic pruritic dermatosis of unknown aetiology can affect males and females of all ages but tends to affect atopic individuals at a younger age.3 Prurigo nodularis can complicate other chronic inflammatory skin diseases such as eczema but can occur in isolation. It can sometimes be associated with hyperthyroidism, chronic kidney disease, coeliac disease, psychiatric disease (e.g. depression, anxiety), haematological malignancy, and viral infections such as human immunodeficiency virus, hepatitis C virus and hepatitis B virus infection.3 The clinical features of prurigo nodularis involve multiple indurated, intensely pruritic papules and nodules that are symmetrically distributed, favouring extensor surfaces, with associated excoriation, crusting and postinflammatory hyperpigmentation.3 Hyperkeratosis, irregular acanthosis, mixed dermal inflammatory infiltrate, increased mast cells and perineural eosinophils are typical histopathological features.3
- Secondary syphilis. Caused by the spirochete Treponema pallidum, secondary syphilis causes a typically nonpruritic, diffuse papulosquamous eruption that develops between three and 10 weeks after inoculation and may be difficult to distinguish from other dermatoses.4 Lesions may be macular, papular or follicular, with a copper-coloured surface, that are symmetrically distributed and involve both mucosal and acral surfaces.4 The painless, indurated chancre of primary syphilis may point to the diagnosis of secondary syphilis. Less than 10% of patients with secondary syphilis will exhibit patchy ‘moth-eaten’ alopecia, and involvement of the oral mucosa is seen in about 20% of patients.4 Mucosal lesions vary in morphology from small superficial ulcers to plaques. Condylomatous lesions may be observed in the anogenital region.4 An accompanying systemic prodrome is common, with symptoms including fever, myalgia, lymphadenopathy, photophobia and headache.4 Dark field microscopic examination of serous discharge will identify the spirochetes in primary syphilis, whereas serological testing is most useful for the diagnosis of secondary syphilis.4 The wide morphological spectrum of syphilis lesions is reflected in the highly variable histopathology, with endothelial swelling, epidermal spongiosis, acanthosis and parakeratosis being the most common findings.4
- Lichen planus (LP). This is the correct diagnosis. LP is an idiopathic inflammatory disease of the skin, nails and mucosal membranes. The characteristic features of classic cutaneous LP are purple/violaceous, planar, polygonal, pruritic papules and/or plaques,5 which are visible in the Figure. Closer inspection reveals a lacy white surface, known as Wickham’s striae, that is often indicative of LP. 6 The disease has a predominance in females, a mean onset in the fifth decade of life for cutaneous LP, and associations with hepatitis C virus seropositivity and autoimmune disorders (e.g. ulcerative colitis, alopecia areata).5 Cutaneous LP most commonly involves the flexural surfaces of the upper limbs, the extensor surfaces of the hands, legs and the presacral skin.5 The oral cavity is the most common site of mucosal disease and may often be the only site of LP. Erosive lesions affecting the anogenital area can produce significant morbidity, resulting in destructive and disfiguring scarring that may require surgical correction.5 Uncommonly, LP may be drug induced. The rash may be indistinguishable from idiopathic LP or present as a nonspecific exanthem or enanthem with an LP-like histopathology. Drugs commonly involved include antimalarials, NSAIDs, thiazides and heavy metals such as gold and mercury (in amalgam). Some supplements and complementary medicines have also been reported to cause LP. Despite the many variants of LP, the histopathological features remain the same: orthokeratosis, thickening of the stratum granulosum, vacuolar degeneration of the stratum basalis and linear band-like lymphocytic infiltration at the dermoepidermal junction.5
Management
The appearance of cutaneous LP warrants a full skin examination to ensure that any other mucosal, anogenital, nail, scalp or ophthalmic LP lesions are identified and treated. Topical and intralesional corticosteroids are first-line treatment for both mucosal and cutaneous LP, with treatment intent to reduce pruritus and restore skin appearance.5 Treatment and monitoring of oral lesions is warranted because there is a 1% risk of development into de novo squamous cell carcinoma.5 Acute LP usually resolves with treatment within 18 months, but patients should be warned that it may persist in a chronic form.6 Other treatment options include systemic corticosteroids, oral retinoids and phototherapy.5
Outcome
This patient had no extracutaneous or anogenital lesions concerning for LP and hepatitis serological testing returned negative results. The diagnosis of LP was confirmed by biopsy. The patient was initially managed with both topical and systemic corticosteroids, but the results were disappointing. Her LP responded very well to acitretin, which she continued regularly for two years until the LP resolved. MT