Genetic testing for familial hypercholesterolaemia: a test for the family

Familial hypercholesterolaemia is a relatively common inherited condition that raises the risk of severe cardiovascular disease 20-fold. Genetic testing to confirm the diagnosis in the index patient followed by GP-led family screening can allow early diagnosis and management to lower risk.
- Genetic testing for familial hypercholesterolaemia (FH) is now listed on the Medicare Benefits Schedule.
- The main benefit of genetic testing for FH is to enable cascade testing of at-risk family members.
- People with FH are at extremely high risk of cardiovascular disease (CVD), and thus have the most to gain from therapeutic intervention and lifestyle advice for CVD risk reduction.
- GPs are ideally placed to support families with FH.
- Cascade testing of family members opens the door for GP-guided diagnosis of family members and management of FH.
- FH is not rare, with a prevalence of about 1 in 300 in Australia; all GPs will encounter FH in their practice.
Familial hypercholesterolaemia (FH) has a prevalence of about 1 in 300, meaning that an estimated 100,000 people in Australia live with FH. Most are unaware of their diagnosis. FH is significantly underdiagnosed and undertreated worldwide. When untreated, men with FH have only a 50% chance of reaching their 50th birthday without serious coronary disease. The burden of premature cardiovascular disease (CVD) in families with FH can be reduced with simple, cost-effective interventions. The greatest benefit is achieved if patients are identified early in life because long-term therapies are most effective.
Genetic testing for FH can support families in confirming the diagnosis of FH and identifying which family members will benefit from interventions to lower their lifetime risk of CVD. FH genetic testing was added to the Medicare Benefits Schedule (MBS) in May 2020 and can be ordered by GPs for screening family members of a person with a genetic diagnosis of FH.
This article reviews the ‘red flags’ that should raise suspicion of FH, the role of genetic testing in diagnosis and family cascade screening, and management of people with FH. The role of the GP is explored.
What is familial hypercholesterolaemia?
FH is an inherited condition affecting the body’s ability to clear LDL cholesterol from the blood. The characteristic elevated LDL-cholesterol levels are present from birth. The consequent lifelong burden of circulating LDL cholesterol on the arteries confers a 20-fold higher risk of severe CVD compared with age- and sex-matched peers. The increased risk is multiplied in the presence of conventional risk factors, such as smoking and diabetes.
FH is inherited in an autosomal dominant pattern, meaning a 50% chance of FH for siblings and children of an affected patient. Thus, when a patient is diagnosed with FH, a whole family is identified as being at risk and should be targeted for screening. FH does not influence triglyceride levels, risk of diabetes, weight gain or obesity.
Family cascade screening
Early identification of people with FH is important for CVD prevention. Population-based algorithms that estimate absolute CVD risk are broadly encouraged for use in adults over 45 years of age, but these calculators are not accurate for people with FH and are applied too late in life. All patients with FH are considered to be at high risk of CVD regardless of age, and interventions should be started immediately, even in young people, to reduce that risk.
Family cascade screening is the most cost-effective method for identifying patients with FH. Cascade screening is the process of screening family members for FH after diagnosis of an initial case (the so-called ‘index patient’). As half of all first-degree relatives and a quarter of second-degree relatives will have FH, cascade screening is a highly effective strategy for identifying patients who will benefit immediately from CVD risk-reducing management.
Screening with LDL-cholesterol tests
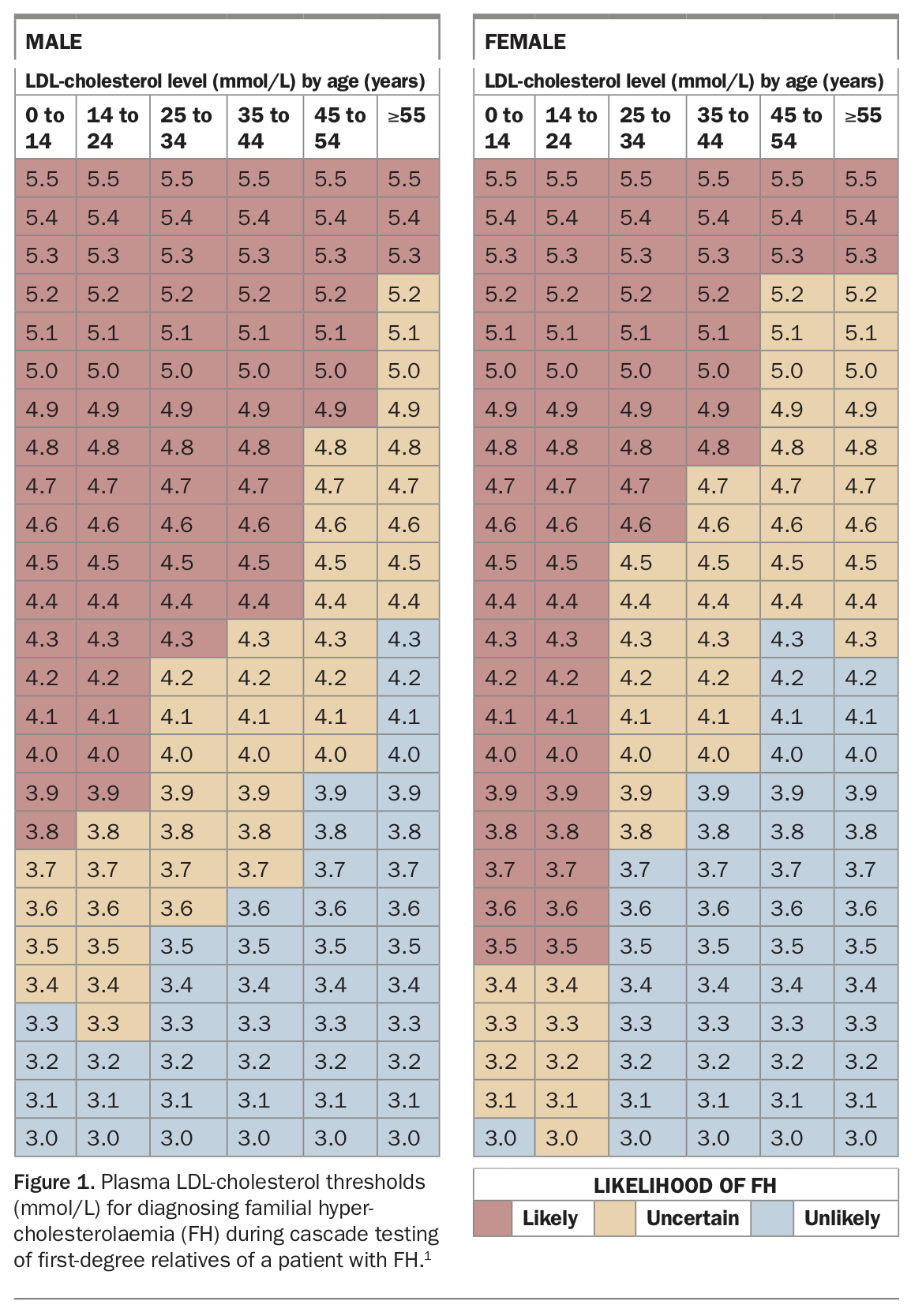
Cascade screening of family members for FH can be done through blood LDL-cholesterol testing. Multiple, consistently elevated results over time are required to confirm the diagnosis (Figure 1).1
{kind=link}
Screening with genetic tests
Because of the confounding factors that influence blood cholesterol levels with increasing age, genetic testing greatly assists cascade testing. When the index patient has a genetic diagnosis, family cascade testing becomes more straightforward. A simple blood sample can confirm or exclude an FH genetic diagnosis in ‘at-risk’ family members.
The US Centers for Disease Control and Prevention (CDC) have long recognised FH as a ‘tier 1’ genomic application, highlighting the many benefits and limited risks of genetic testing in families with FH. In Australia, FH genetic testing was added to the MBS in May 2020. Guidance on the care of people with FH in Australia was recently updated, and recommendations relevant to genetic testing are shown in Box 1.2
{kind=link}
The MBS now allows GPs to order cascade genetic testing for first- and second- degree relatives after more comprehensive testing has identified a causative genetic variant in the family index patient. This opens the door for GP-guided diagnosis and management of affected family members. Only the more comprehensive genetic testing of the index patient requires specialist involvement.
How can we recognise FH in our patients?
GPs are ideally placed to identify patients who may have FH, given the likely 100,000 Australians who have not yet been diagnosed. Although the spectrum of signs and symptoms of FH can vary widely, patient features that should prompt consideration of FH include:
- premature atherosclerotic cardiovascular disease (men aged under 55 years, women under 60 years)
- a family history of premature cardiovascular disease
- LDL-cholesterol level over 5 mmol/L, particularly in the first four decades of life
- tendon xanthomas, xantholasmas or corneal arcus in the patient or a family member.
Tendon xanthomas are found on examination in about 20% of people with FH, so cannot be relied on to identify all cases. However, as they are fairly specific to FH, if a tendon xanthoma is seen in combination with a high LDL-cholesterol level, FH can be diagnosed with reasonable confidence.
There is significant overlap between the cholesterol profiles of the average population and patients with FH. The possibility of FH should be considered in any patient with an LDL-cholesterol level above 5 mmol/L in the absence of a clear secondary cause (Box 2).3
{kind=link}
The next steps are to:
- clarify whether other causes of elevated cholesterol levels have been ruled out, including hypothyroidism, nephrotic syndrome and a high-fat diet (Box 2)3
- ensure at least two measurements of LDL-cholesterol level are obtained
- clarify any family history of high cholesterol or CVD
- ascertain whether FH genetic testing has already been performed in a family member. The result may allow the patient to have genetic testing using simpler, less expensive laboratory procedures associated with cascade screening.
Where does genetic testing fit?
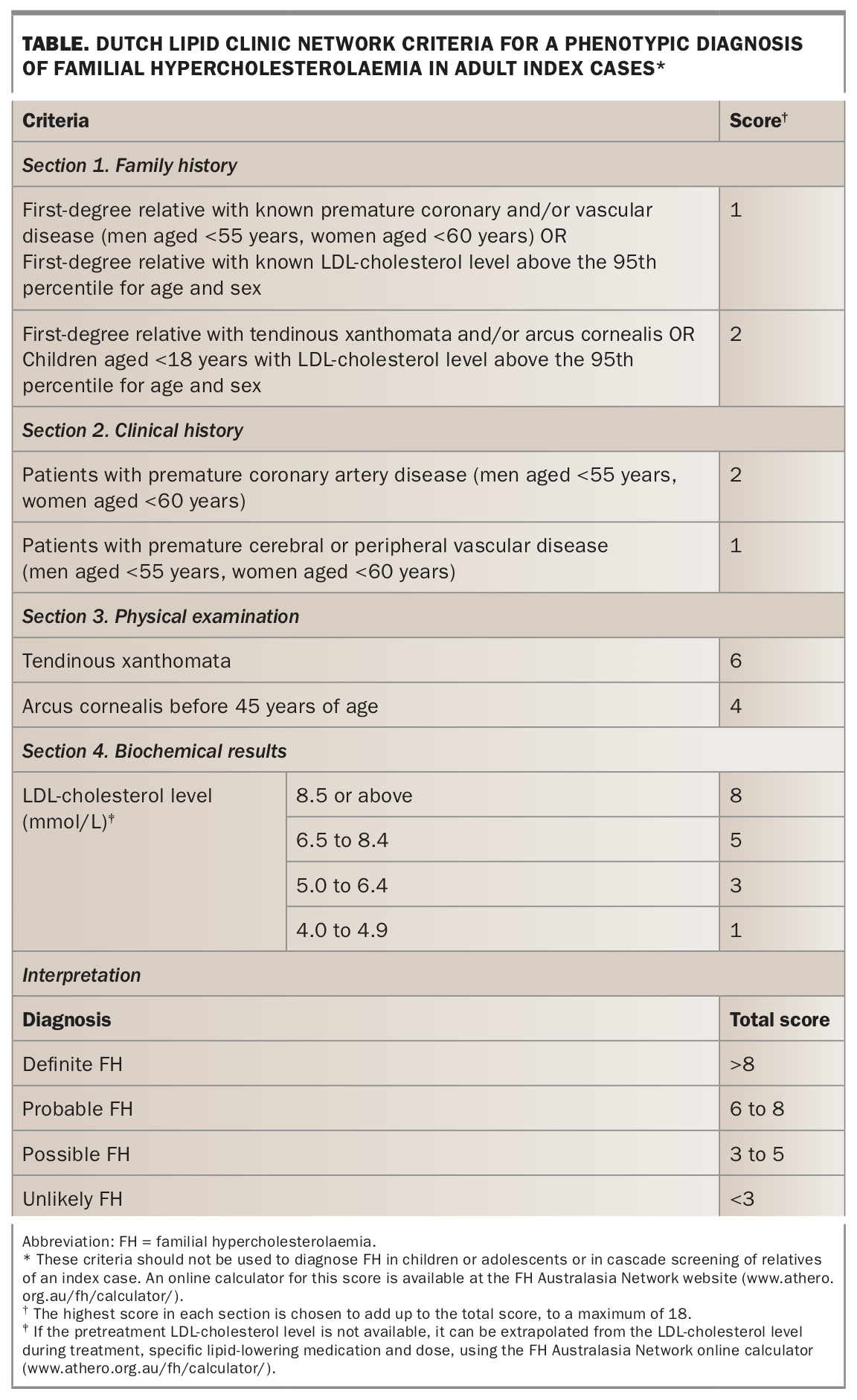
A genetic test is not required to diagnose FH but should be offered to confirm a clinical diagnosis when possible.2 However, a clinical diagnosis is sufficient to provide appropriate management advice and treatment to an individual. The criteria of the Dutch Lipid Clinic Network (DLCN) for a phenotypic diagnosis of FH are shown in the Table.
{kind=link}
The greatest benefit of a genetic test result is to enable family cascade testing to identify relatives who could benefit from specific advice on CVD risk reduction. Not all patients will choose to confirm the diagnosis with genetic testing, but their first- and second-degree relatives should have LDL-cholesterol screening regardless of a genetic diagnosis in the family.
Genetic testing of the LDLR, APOB and PCSK9 genes confirms a diagnosis of FH in about 80% of cases. However, testing is not sufficiently sensitive to exclude a diagnosis of FH in the case of a negative genetic result. Clinical assessment is still essential for diagnosis and to inform relatives of their risk of FH.
Fortunately for those with a negative genetic test result, PBS access to novel cholesterol-lowering therapies is no longer restricted to those with a genetic diagnosis. A genetic result is not required to access treatment deemed necessary and approved through authority prescription.
The MBS currently offers a rebate for the first broad FH genetic test in a family, ordered by a specialist. After a specific gene variant has been identified, the MBS supports both GP- and specialist-ordered cascade gene testing for this variant in first- and second-degree relatives. A suggested algorithm for family cascade screening for FH is shown in the Flowchart.
If broad genetic testing for FH in an individual has negative results then cascade gene testing is not available for relatives. Instead, LDL-cholesterol screening and clinical assessment of at-risk relatives should be performed to diagnose FH. The DLCN criteria should not be used for cascade screening. Blood LDL-cholesterol testing is relied on to make a diagnosis in relatives after FH is confirmed in one individual in a family (Figure 1). Secondary causes of high LDL-cholesterol levels need to be considered, and multiple LDL-cholesterol results should be reviewed over time to confirm the diagnosis.
Genetic test results and variant classification
The first genetic test arranged in the family must be broad and examine all three of the well characterised genes known to cause FH: the LDLR, APOB and PCSK9 genes. On sequencing these genes, the laboratory classifies any identified gene variants according to how likely they are to contribute to disease. Most commonly variants are classified on a spectrum, such as ‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’ and ‘benign’. This classification is dynamic, with new evidence on the complexities of the genes being published continually.
Cascade genetic testing in relatives can be offered for variants classified as pathogenic or likely pathogenic, as there is a high level of confidence that these variants are the cause of FH in the family. Testing for a pathogenic or likely pathogenic variant in relatives at risk of FH provides clear information on whether or not they have inherited FH.
There is much normal variation in the LDLR, APOB and PCSK9 genes, and many variants are known to be benign and not associated with an increased LDL-cholesterol level. Testing for benign variants in relatives is not appropriate.
A variant of uncertain significance or ‘VUS’ may be reported if there is insufficient evidence to confirm its causal role in high cholesterol levels. In this case, a laboratory may not be able to determine accurately whether the gene variant causes FH or simply represents natural variation. A VUS cannot be used for cascade testing in relatives to diagnose FH as its presence or absence would not confirm or exclude, respectively, a diagnosis of FH.
Over time, we hope that further evidence will accrue to reclassify a VUS as pathogenic or benign. If a patient has a VUS result, referral to a genetics clinic might be considered. Genetic counsellors can help facilitate testing of other relatives to track a VUS in the family and liaise with a laboratory for family-specific VUS reclassification.
If a family gene variant is not identified, cascade screening and FH diagnosis in relatives relies on repeat LDL-cholesterol results.
GP-led family cascade screening
Over 1000 different gene variants are known to cause FH. Knowing the specific gene variant causing FH in a family is crucial for cascade genetic testing in relatives. After the genetic variant causing FH in a family is identified, Medicare-funded cascade genetic testing of first- and second-degree relatives can be ordered by a GP or specialist, and should be offered to all relatives, irrespective of cholesterol level.2
Consent should be documented for all patients who undergo genetic testing. National unifying consent forms for genetic testing are being rolled out to support the increase in genetic testing in a wide range of specialties (www.australiangenomics.org.au/resources/for-professionals/national-clinical-consent/). Primary prevention patients may wish to seek information about potential insurance implications before genetic testing. Genetic testing for FH should always be done alongside clinical evaluation of LDL-cholesterol levels.
When ordering a cascade gene test for a patient, the laboratory must be supplied with details of the known gene variant causing FH in the family. This is most commonly achieved by sending the blood sample for cascade gene testing to the same laboratory that performed the first genetic test in the family. Providing a copy of the original genetic test report to the testing laboratory can be helpful, or details such as patient name or laboratory reference number from the original report can be stipulated on the request form. The genetics pathologist needs to know how the current patient is related to the previously tested relative (e.g. sibling, child or parent). Most importantly, the pathologist needs to know the specific gene variant details, so as to focus on that variant and not reanalyse the entire gene panel. Tips for ordering a cascade gene test for FH are shown in Box 3.
{kind=link}
Management and monitoring of patients with FH
Lifelong adherence to lifestyle changes and medications are paramount to reducing CVD risk in people with FH. It is essential for individuals and families to be educated and engaged in shared decision-making regarding their medical care. Empowered patients who are actively involved in management decisions are more likely to take charge of their own personalised CVD risk-reducing interventions and adhere to lifelong changes. Management strategies will of course change over time, with age, potential comorbidities and life stage. A regular GP is ideally placed to actively identify changes in circumstances and help patients to tailor their treatment.
LDL-cholesterol testing remains the most important measure for guiding treatment. Counselling about lifestyle factors and diet are essential at all ages as these interventions are likely to benefit the entire family. After diet and lifestyle factors have been optimised, patients with FH usually continue to have high levels of LDL cholesterol because of the inherited gene variant affecting their body’s LDL- cholesterol regulation.
Thus, medical therapies to lower LDL cholesterol should be considered for all adults on diagnosis of FH as gold standard primary prevention. Patients may wish to trial intensive diet and lifestyle strategies for a predetermined time frame, such as three to six months, to see how low they can reduce their cholesterol levels, before reviewing levels again with their GP and initiating lipid-lowering therapy. Therapies should also be considered on a case-by-case basis for children and adolescents, starting around 10 years of age, with the assistance of a paediatric specialist.
Patients diagnosed with FH on cascade testing by their GP can continue to be treated as per standard lipid-lowering therapy guidelines. Referral to a specialist (a lipidologist, endocrinologist or cardiologist) may be considered for more complex cases, such as in the setting of secondary prevention, multiple comorbidities or intolerance to standard lipid-lowering therapies. Risk assessment for FH patients should also include measurement of the independent risk factor lipoprotein(a), which, like LDL cholesterol, is commonly elevated in the presence of FH.
The case history of a family with FH is described in Box 4. This case highlights the benefits of early diagnosis of FH, enabling early commencement of primary management to reduce the risk of premature CVD.
{kind=link}
Resources on FH for GPs and patients
Many resources are available to support patient and GP education about FH. These include:
- support for the consent process for genetic testing (www.australiangenomics.org.au/resources/for-professionals/national-clinical-consent/)
- written information and contact details for referring to
– public genetics clinics (Centre for Genetics Education, www.genetics.edu.au/genetic-services/general-genetics-clinics)
– specialists with an interest in treating FH (FH Australasia Network, www.athero.org.au/fh/health-professionals/fh-specialists/).
The detection and management of FH is informally monitored through a national FH registry, which provides helpful feedback on the status of FH in Australia. It is highly desirable that most definite FH cases, particularly those with a known causative genetic variant, are recorded on this voluntary registry (www.athero.org.au/fh/patients/fh-registry/).
Conclusion
Cascade screening of relatives of people with FH facilitates early diagnosis of FH in a population that is otherwise unaware of their condition. Early diagnosis is crucial as it enables early management of cholesterol levels and reduces the burden of severe, premature CVD.
Genetic testing is not essential for cascade screening in at-risk relatives but provides a useful resource for the family. The MBS now allows GPs to order cascade genetic testing in first- and second-degree relatives after a confirmed genetic diagnosis of FH in a family member. This opens the door for GP-guided diagnoses and family-centred management. MT