Inherited arrhythmia syndromes: caring for families in 2021

Dr Isbister is a Cardiologist and PhD candidate at the Agnes Ginges Centre for Molecular Cardiology, Centenary Institute and The University of Sydney, Sydney; a PhD candidate in the Faculty of Medicine and Health, The University of Sydney; and a Research Fellow in the Department of Cardiology, Royal Prince Alfred Hospital, Sydney.
Professor Semsarian is Head of the Agnes Ginges Centre for Molecular Cardiology, Centenary Institute and The University of Sydney, Sydney; Professor of Medicine in the Faculty of Medicine and Health, The University of Sydney; and a Senior Staff Specialist Cardiologist in the Department of Cardiology, Royal Prince Alfred Hospital, Sydney, NSW.
Arrhythmia
Congenital heart defects

Inherited arrhythmia syndromes are an important cause of sudden cardiac death in young people. Although the individual conditions in this group have unique features and management strategies, an awareness of shared clinical red flags along with family screening can help prevent sudden cardiac death.
- Early identification of patients at risk of inherited arrhythmia syndromes (IAS) is a crucial step in reducing sudden cardiac death in our community.
- Electrocardiographic signatures of IAS are dynamic, and provocation testing should be considered when clinical suspicion of an IAS is high.
- Understanding of the genetic basis of IAS continues to evolve, and patients’ genetic testing results should be reassessed periodically.
- Lifestyle modifications are key to reducing arrhythmic events in patients with IAS.
- Care for patients and families with IAS is optimally provided by a multidisciplinary genetic heart disease team.
The inherited arrhythmia syndromes (IAS) are traditionally grouped together on the basis of their shared pathophysiology of cardiac ion channel disruption and common clinical endpoint of malignant ventricular arrhythmia. These conditions exhibit a broad spectrum of disease, from incidental diagnosis in an asymptomatic individual to sudden cardiac death (SCD). Indeed, up to 40% of SCD victims under 35 years of age have no cause identified on autopsy, and primary cardiac arrhythmia is presumed to be the culprit.1 Key challenges for clinicians include the identification of at-risk patients, accurate risk stratification and timely management aimed at preventing SCD events. Rapidly evolving knowledge of the genetic basis of these conditions continues to provide insights into their unique clinical features, which can be used to improve diagnosis and optimise therapeutic strategies.
This review aims to provide busy clinicians with practical information that can help them care for patients or families at risk of an IAS. We will highlight the clinical situations where IAS should be considered and discuss the principles of diagnosis, utility of genetic testing and importance of risk stratification in optimising management and preventing SCD events. Finally, we will provide a brief update on the specific features of three of the more common IAS.
Clinical presentation
Although IAS are rare, the diverse presentations of patients with, or at risk of, a primary inherited arrhythmia requires clinicians to be aware of the ‘red flags’ that should prompt further assessment. Clinical situations where IAS should be considered are summarised in Box 1.
{kind=link}
In individuals presenting with possible IAS symptoms, such as syncope, a detailed history is of great importance. For example, understanding the circumstances in which a syncopal event occurred allows the clinician to identify features that may favour a diagnosis of cardiac arrhythmia over a simple vasovagal event. Syncope during exercise should always raise suspicion of a cardiac cause. Certain triggers, such as swimming, emotional stimulus or a loud noise, can even provide a clue towards a specific IAS (see below). Individuals who have experienced recurrent syncopal events or those with a history of seizures but normal results on neurological assessment or whose condition is refractory to therapy should undergo detailed cardiac investigation.
Enquiring about a family history of IAS or SCD is also key, and where elicited should heighten suspicion and guide investigations. A family history of SCD or known IAS is clearly concerning, but clinicians should also seek a more obscure family history that might indicate a risk of IAS. This includes any deaths due to sudden infant death syndrome, unexplained motor vehicle accidents or near- drowning (particularly in the young), or seizures, especially if refractory to therapy.
In some people, the first symptom of an IAS is SCD. Consequently, an asymptomatic individual with an incidentally detected ECG abnormality requires timely cardiac evaluation.
Patients may seek screening for an IAS after an IAS diagnosis is established in their family or after the sudden death of a relative where no cause is identified on autopsy. Resources that may assist in this situation are listed in Box 2.
{kind=link}
Diagnosis and management
The individual conditions encompassed under the ‘IAS’ banner have unique clinical features, ECG signatures, genetic drivers and targeted therapeutic options. However, there are some shared principles of diagnostic evaluation, management approach and SCD prevention that can help clinicians care for those at risk of these conditions.
Team-based approach
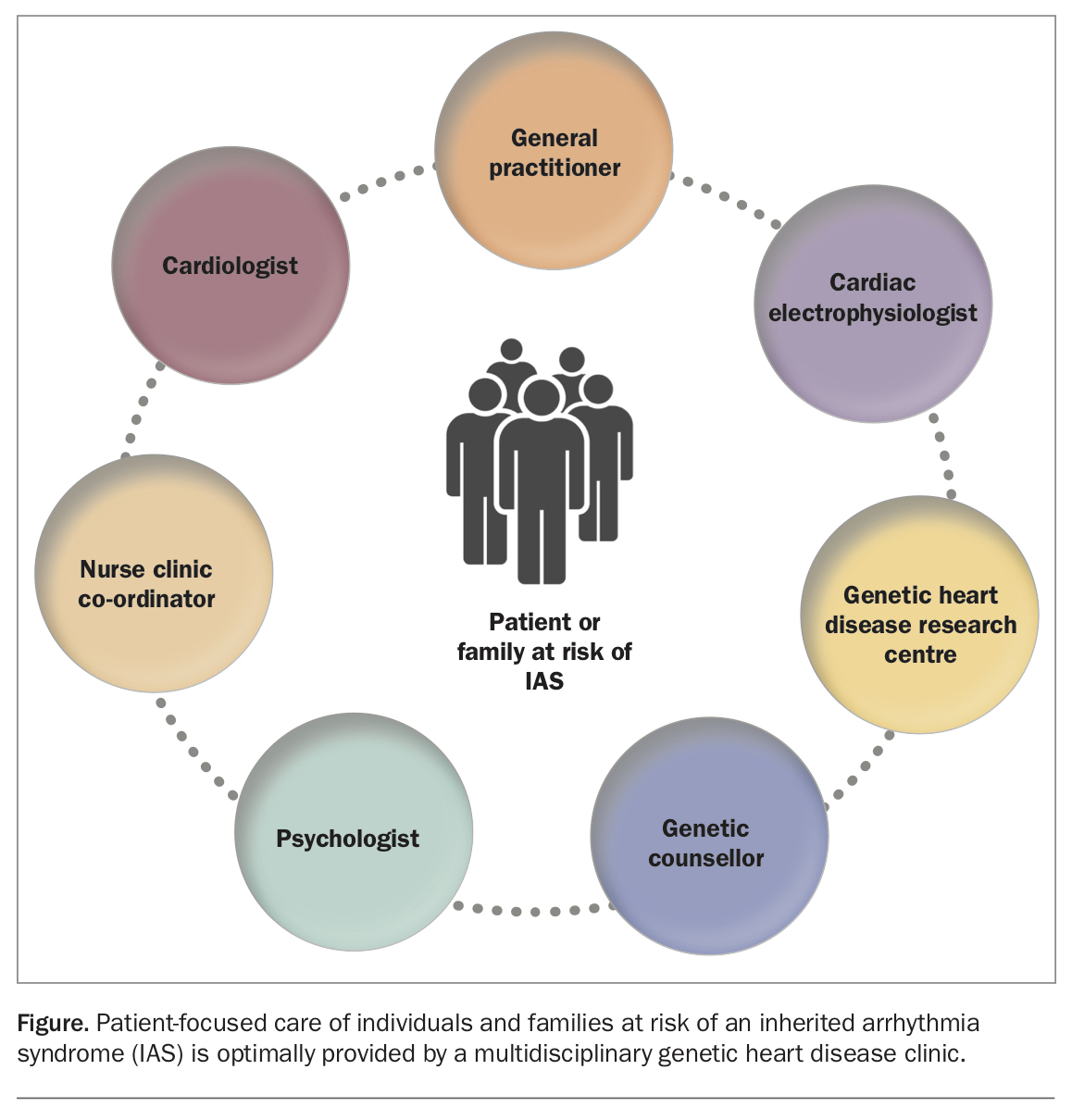
Patient-centred care for individuals and families with an IAS is optimally provided by a multidisciplinary team (Figure). GPs are key in identifying at-risk patients and are often well positioned to engage family members in screening when a diagnosis is confirmed. Cardiologists, cardiac electrophysiologists and genetic heart disease specialists all play important roles in detailed phenotyping, risk stratification and guiding management.
{kind=link}
Expert genetic counselling, ideally by a specialised genetic counsellor, ensures patients can consider the potential implications of both testing and results, before any genetic testing. The importance of this must not be underestimated.
Linking patients with teams based at IAS research centres empowers families who are keen to participate in the global effort to improve understanding and management of these rare conditions.
Provocation testing
The ECG abnormalities of IAS are dynamic and can vary with circadian rhythm and activity, as well as certain medications and the metabolic milieu. A normal resting 12-lead ECG does not exclude the diagnosis of an IAS, and provocation testing may be employed to uncover concealed abnormalities when clinical suspicion of an IAS is high. Provocation testing may be useful in:
- individuals with a family history of an IAS
- patients who have survived an unexplained cardiac arrest
- patients who have nondiagnostic ECG abnormalities at rest but have symptoms with exertion or an emotional stimulus.
These tests exploit knowledge about the driving pathophysiology of the specific IAS or their clinical triggers and are outlined in the Table.2-8
{kind=link}
Genetic analysis
Genetic testing can help in the care of patients and families with, or at risk of, IAS in three main circumstances, as follows.
Patient diagnosed with IAS
First, genetic testing is recommended in individuals with a confirmed diagnosis of an IAS.9 Identification of a pathogenic (disease-causing) genetic variant can support a diagnosis, aid in risk stratification and allow for genotype-directed therapy where available. It can even be used to reduce the risk of the condition being passed on to the next generation through preimplantation genetic diagnosis.
Family of patient with a gene variant
In families where a gene variant causing an IAS has been identified, relatives can be screened for the specific genetic change (cascade testing), in addition to routine clinical screening. Relatives with the variant can then be monitored or treated as appropriate, whereas those without the variant can be reassured and released from long-term clinical follow up.
Following sudden cardiac death
In cases of SCD where no abnormalities are identified at autopsy, cardiac arrhythmia is the presumed culprit. Postmortem genetic testing can identify a cause of death in up to 20% of people with SCD.1,10 Most pathological variants identified are in IAS-associated genes. Identifying the cause of death in this setting provides families with answers about their relative’s death and aids in family screening aimed at preventing further SCD events in the family.
Interpretation of genetic results
Despite an explosion of knowledge about the role of genes in disease over recent decades, understanding of the genetic architecture of IAS continues to evolve. Determining whether an observed variant is the cause of disease, as opposed to a benign genetic change, requires expert examination of evidence from population databases of human genetic variation, computer modelling to predict the likely in vivo effect and evidence of the same change in other affected individuals around the world.
The yield of genetic testing varies significantly between the different IAS, and the absence of an identifiable cause on genetic testing does not discount an established clinical diagnosis. It is important to remember that the understanding of a specific variant’s role in disease can change over time, and periodic reassessment of both positive and negative genetic results with the potential to impact on care of those with an IAS is crucial.9
Management and risk stratification
The cornerstone of care for all patients with an IAS is education on lifestyle modifications to avoid arrhythmia triggers. The use of pharmacotherapy varies significantly between the conditions, from universal use (e.g. beta blocker therapy in patients with catecholaminergic polymorphic ventricular tachycardia [CPVT]), to only rarely indicated (e.g. quinidine in Brugada syndrome).
Implantable cardioverter defibrillators (ICDs) are used in patients considered to be at increased risk of SCD. ICD use is limited by device-related complications, especially in this relatively young cohort of patients who may expect to live with these devices for many years. Syncope in the setting of a primary arrhythmic syndrome is an important indicator of arrhythmia risk.5 However, other factors considered in risk stratification are syndrome-specific and are discussed below.
Features of specific IAS
The specific clinical features, arrhythmia triggers, risk modifiers and genetic associations and management options of the three most common IAS are discussed below and summarised in the Table.2-8
Familial long QT syndrome
Long QT syndrome is a disorder of cardiac repolarisation that predisposes to the development of a polymorphic ventricular tachycardia known as torsades de pointes, which can degenerate into ventricular fibrillation.
Diagnosis of long QT syndrome
Long QT syndrome is diagnosed:
- based on the Schwartz score, a criterion that includes the 12-lead ECG findings as well as clinical features and family history to ascertain the probability of LQTS11
- in the presence of a QT interval corrected for heart rate (QTc) over 500 ms alone
- in the presence of an unequivocally disease-causing variant in a gene implicated in long QT syndrome.5
In patients who are considered at risk but have a nondiagnostic resting ECG, provocation testing can be useful to confirm or exclude the diagnosis. The ‘quick standing’ test exploits the relatively slow QT response to an abrupt increase in heart rate after standing to expose a long QTc.12 QT response to adrenergic stimulus can be assessed by an exercise stress ECG, measured at four minutes into the recovery phase, or, when an exercise ECG is not possible, by an adrenaline challenge.13,14
The genetic cause of long QT syndrome can be identified in up to 60% of cases.15 Although 17 genes have been associated with long QT syndrome, the vast majority of pathological variants identified reside in just three of these genes:
- KCNQ1 (potassium voltage-gated channel subfamily Q member 1 gene) – termed long QT syndrome type 1
- KCNH2 (potassium voltage-gated channel, subfamily H [eag-related], member 2 gene) – long QT syndrome type 2
- SCN5A (sodium voltage-gated channel alpha subunit 5 gene) – long QT syndrome type 3.
Identifying a genetic variant is particularly useful in patients with long QT syndrome. In addition to assisting in diagnosis and care of relatives, it informs risk stratification and allows genotype-specific therapy.
Management of long QT syndrome
All patients with a diagnosis of long QT syndrome must avoid QT-prolonging medications (listed at crediblemeds.org) and any genotype-specific triggers where possible. Beta blockers are recommended in patients with symptoms and are also used routinely in all but the lowest risk patients.5 ICD therapy is recommended in high-risk patients and those with recurrent events while taking beta-blockers. Left sympathetic cardiac denervation surgery is an important treatment adjunct in patients with long QT syndrome, particularly in those with recurrent events on standard therapy and those at high risk who have a contraindication to an ICD.5
Brugada syndrome
Historically, Brugada syndrome has been considered a channelopathy, but there is ongoing conjecture about the underlying pathophysiology. Indeed, the syndrome is now hypothesised to be a focal form of cardiomyopathy.16
Diagnosis of Brugada syndrome
Brugada syndrome is defined by the classic ‘type 1’ ECG signature (ST-segment elevation of 2 mm or more with coved-type morphology in one or more of the right precordial leads; Table). A spontaneously occurring type 1 ECG pattern is sufficient for diagnosis. However, when the type 1 pattern can be provoked only by fever or drugs, it has been recommended that additional clinical or genetic criteria are considered before conferring a diagnosis.17
A single genetic cause of Brugada syndrome is identified in only 20% of patients, in the SCN5A gene. Evidence is growing that an accumulation of genetic risk modifiers may account for a significant portion of disease.
Management of Brugada syndrome
Lifestyle modifications such as fever management and avoidance of drugs known to provoke arrhythmia (listed at brugadadrugs.org) are recommended for all patients with Brugada syndrome. The risk of SCD in asymptomatic patients is low, in the order of 1% per year. ICD therapy is recommended only in those who have experienced symptoms (such as syncope, cardiac arrest, nocturnal agonal breathing) or documented ventricular arrhythmia.3
The role of adjunctive therapy such as quinidine or catheter ablation continues to evolve. However, it is currently largely reserved for those who have recurrent arrhythmias and those who have an elevated risk of SCD and a contraindication to ICD therapy.
Catecholaminergic polymorphic ventricular tachycardia
CPVT is a rare IAS that usually presents in childhood or adolescence. It is highly lethal when untreated, and this is reflected by the over-representation of the condition in SCD cohorts.10,18 Syncope during adrenergic stimulation is the cardinal feature of CPVT, and events typically occur during exercise or emotional stress. Recent reports have identified that excitement during electronic games can uncover CPVT, and syncope occurring in this setting should prompt cardiac assessment.19,20
Diagnosis of CPVT
The resting ECG is normal in people with CPVT, and ventricular arrhythmias become apparent only during exertion or excitement, usually captured during a stress test or on 24-hour Holter monitoring. Diagnosis is confirmed when ventricular arrhythmia, classically bidirectional ventricular tachycardia (VT) but also polymorphic ventricular ectopy or polymorphic VT, is documented during exercise or adrenergic stimulation in the absence of structural heart disease or other identifiable causes in those aged under 40 years.5 In those aged over 40 years, coronary disease and other causes should be excluded before diagnosis.
Identification of a pathogenic variant is sufficient for diagnosis of CPVT.5 A genetic variant causing CPVT can be identified in around 65% of cases, most commonly in the ryanodine receptor gene (RYR2) but also in the calsequestrin gene (CASQ2).21
Management of CPVT
As with all IAS, lifestyle modifications to avoid triggers are crucial in patients with CPVT, including strenuous exercise and any known emotional triggers. Given the high mortality rate and poor risk stratification available, beta-blocker therapy is recommended in all patients with a diagnosis of CPVT, including those without symptoms.
In patients with recurrent arrhythmia on optimally titrated beta-blocker therapy, adjuncts such as flecainide or left cardiac sympathetic denervation can be considered. ICD therapy is avoided where possible as shocks can drive adrenergic stimulation and precipitate further arrhythmia.
Conclusion
Early identification of patients at risk of an IAS is a crucial step in reducing the tragedy of SCD in our community. Awareness of the presentations that should prompt consideration of these rare conditions, as well as the principles of diagnosis and the role and challenges of genetic testing, will help clinicians care for these patients and their families. MT
COMPETING INTERESTS: None.
Acknowledgements: Dr Semsarian is the recipient of a National Health and Medical Research Council Practitioner Fellowship (no. 1154992).