Pulmonary hypertension – when to suspect and refer

Pulmonary hypertension is an umbrella term that may describe either a primary, progressive, devastating disorder of the pulmonary vascular system or a complication of an array of medical conditions. Early detection of the former is crucial, as effective treatment is available and can avert an otherwise poor prognosis for these patients.
- The main symptoms of pulmonary hypertension (PH) are progressive dyspnoea, fatigue and exercise intolerance.
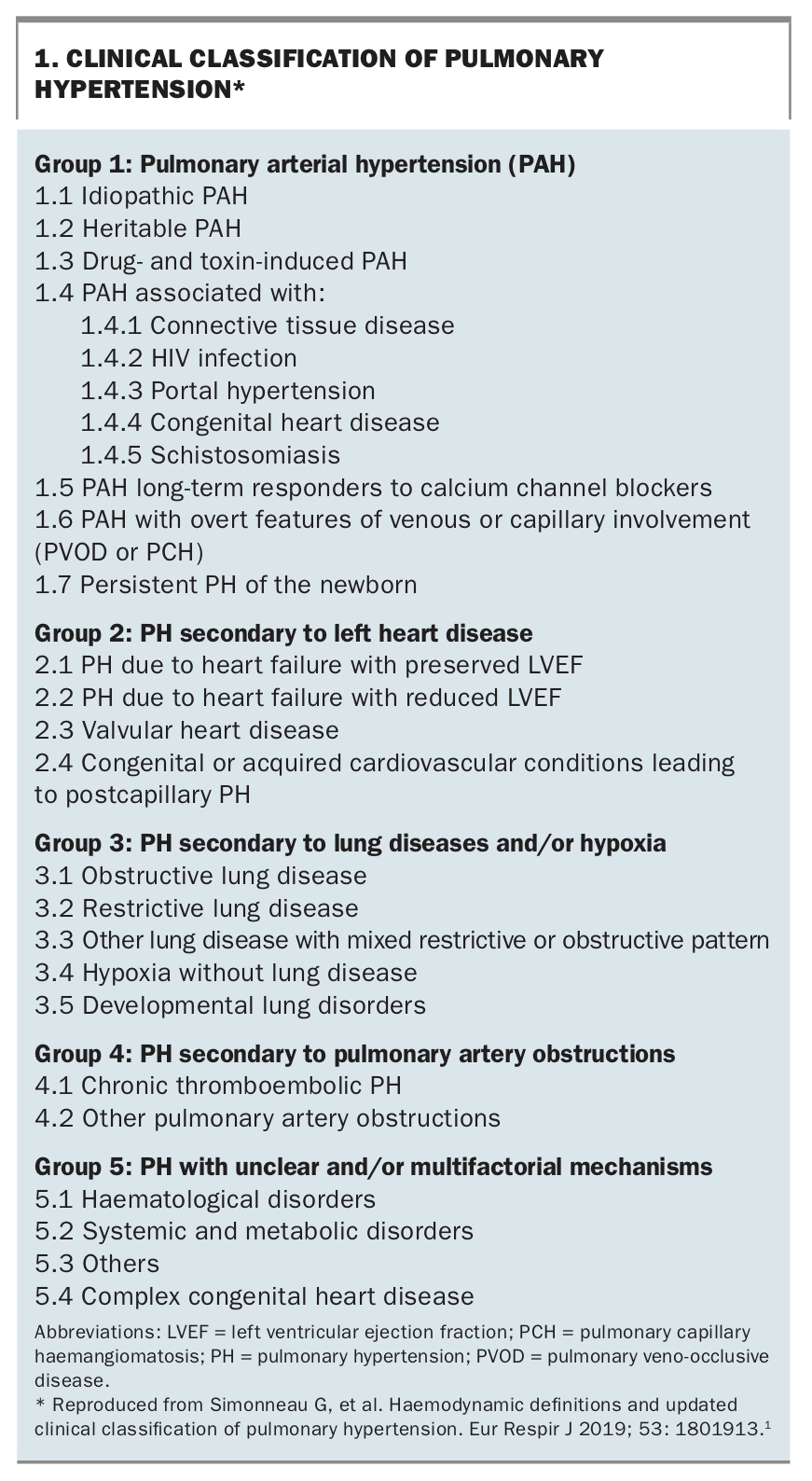
- PH can be associated with various cardiac, respiratory and other systemic disorders and is classified into five groups to help streamline treatment decisions.
- Diagnosis of the most severe forms of PH is often delayed because of its nonspecific symptoms, leading to detection at a more advanced stage of disease.
- Expert PH centres specialise in managing patients with Group 1 (pulmonary arterial hypertension) and Group 4 (chronic thromboembolic pulmonary hypertension) disease.
- Patients with Group 2 or 3 PH (secondary to left heart disease or chronic lung disease) are usually managed primarily by a cardiologist or respiratory physician, respectively, although select patients in these groups may also benefit from review in an expert PH centre.
Pulmonary hypertension (PH) refers to a range of disorders with often nonspecific symptoms and clinical presentations but united by the common finding of elevated pressure in the pulmonary circulation. Given the heterogeneity of these conditions, the various forms of PH have been classified into five groups to streamline therapeutic decision making (Box 1).1 This review aims to highlight when PH should be suspected in patients presenting to primary care and to provide an updated overview of the condition and the general principles of diagnosis and management.
{kind=link}
When to suspect pulmonary hypertension
Although progressive exertional dyspnoea is the most frequently reported symptom of PH, the presentation is often nonspecific and insidious. Fatigue or exercise intolerance may be more prominent, whereas some patients report cough, lightheadedness or chest discomfort. With more advanced disease, patients may experience syncope or symptoms related to right heart failure, such as ankle swelling or abdominal distension.
Physical examination might be essentially normal in the early stages of disease. Over time, patients may develop jugular venous distension, a parasternal heave, loud second heart sound (P2 component), murmurs of tricuspid or pulmonary regurgitation, peripheral oedema, ascites, hepatomegaly or splenomegaly. Physical signs of conditions associated with PH, such as systemic sclerosis, congenital heart disease or cirrhosis, may also be apparent.
Diagnosis
Investigation of dyspnoea of unknown cause is beyond the scope of this article, but it is important to understand that standard investigations may yield apparently normal or nonspecific results in patients with PH, particularly in its earlier stages. Patients with relevant underlying diseases may have pertinent findings on imaging (e.g. cardiomegaly or hyperinflated, emphysematous lungs) or spirometry (e.g. airflow obstruction). However, transthoracic echocardiography is the most important noninvasive screening tool for PH and should be strongly considered when investigating dyspnoea without clear cause. For many patients, evidence of PH may be an incidental or unexpected finding on echocardiography performed for an unrelated indication. An overview of the diagnostic approach to suspected PH is shown in the Flowchart.2
Several echocardiographic findings point towards PH – an elevated estimated right ventricular systolic pressure (RVSP) based on tricuspid regurgitation velocity is the most well recognised, but other indicators include flattening of the interventricular septum and right atrial or ventricular dilatation. Echocardiography also provides invaluable information about left heart function and valvular disease, which may be implicated as the underlying cause of PH. Recent data from the National Echocardiography Database Australia suggest that an estimated RVSP of 30 mmHg may represent the upper limit of normal, in contrast to the higher thresholds accepted previously.3 Patients with an estimated RVSP above 30 mmHg appear to be at greater risk of all-cause and cardiovascular mortality, independent of the presence of left heart disease, age or body mass index.
Right heart catheterisation is the invasive means of confirming PH, which is characterised by an elevated resting mean pulmonary arterial pressure above 20 mmHg, although this finding alone can result from pulmonary vascular disease, left heart disease, left-to-right cardiac shunts or numerous other causes. The accepted mean pulmonary arterial pressure cut-off has recently been reduced from 25 mmHg, in view of mounting evidence of worse outcomes with pressures in excess of the lower threshold of 20 mmHg.1 This change may facilitate identification of at-risk patients at an earlier stage of disease, thereby allowing earlier intervention.
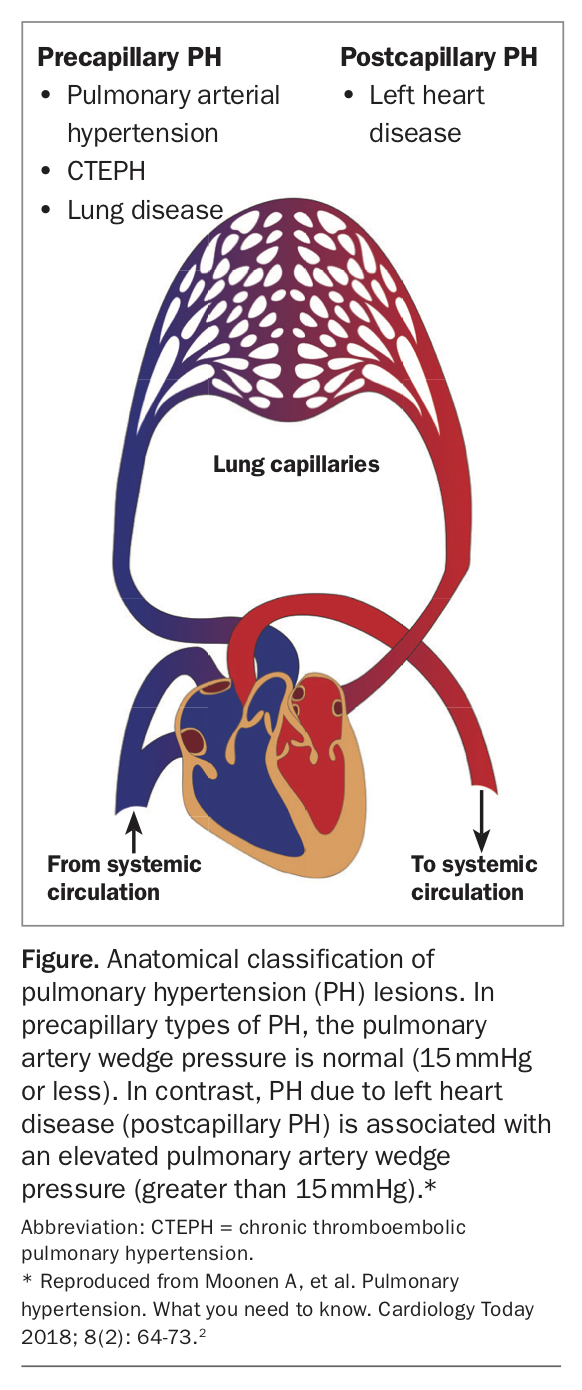
Further, right heart catheterisation allows PH to be characterised as precapillary or postcapillary in origin (Figure),2 based on the pulmonary capillary or pulmonary arterial wedge pressure (PAWP), which has important implications for disease aetiology and therapeutic options. Precapillary PH suggests pulmonary vascular disease and is indicated by pulmonary vascular resistance of greater than or equal to 3 Wood units and PAWP less than or equal to 15 mmHg. It includes Groups 1, 3 and 4 and some patients in Group 5.1 A PAWP above 15 mmHg reflects raised left atrial pressure and implies postcapillary PH, in which left heart disease drives the elevated pulmonary pressures (at least in part).
{kind=link}
Classification of pulmonary hypertension
Group 1. Pulmonary arterial hypertension
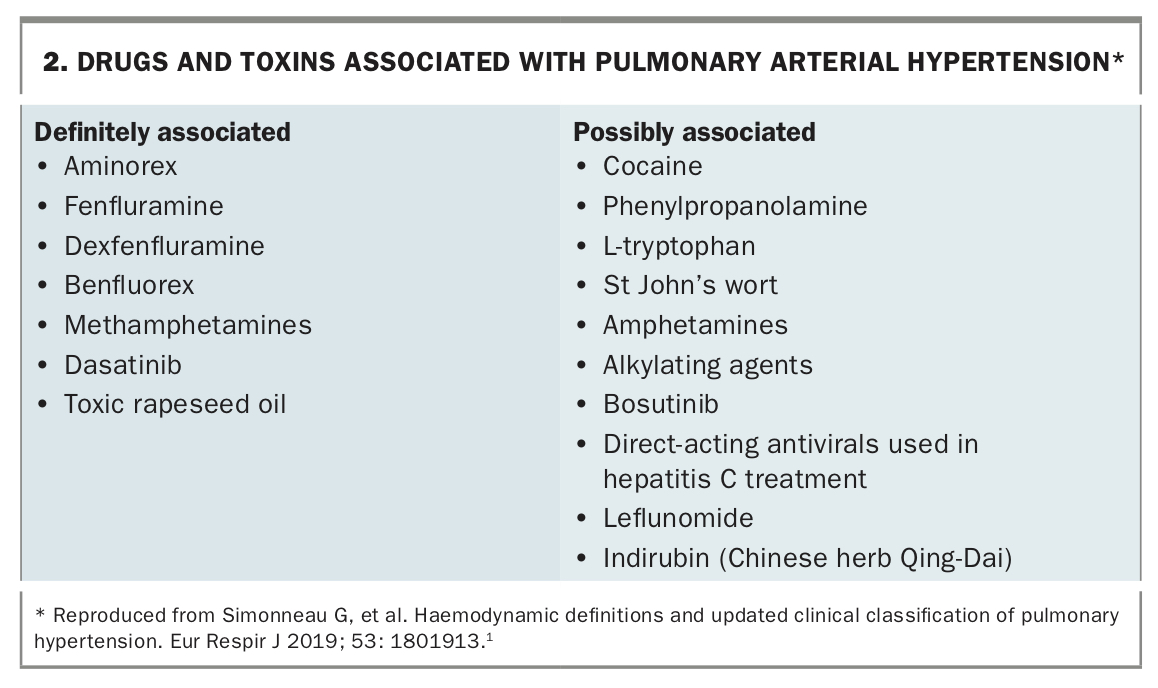
Pulmonary arterial hypertension (PAH) encompasses a relatively rare group of conditions (Box 1), with an estimated prevalence of 15 per 100,000 and an annual incidence of 10 to 15 per million people.4 About half of these people will have idiopathic, heritable or drug- and toxin-induced PAH.5 Idiopathic PAH is more likely to affect younger and middle-aged women. Several genetic abnormalities have been associated with heritable PAH, with mutations in BMPR2 being the most common.6 Drug- and toxin-induced PAH has been linked to the use of amphetamines, methamphetamines, several anorexigens that have been withdrawn worldwide and other substances (Box 2).
{kind=link}
PAH is often associated with connective tissue disease, particularly systemic sclerosis or systemic lupus erythematosus, and less so with rheumatoid arthritis, dermatomyositis or Sjogren’s syndrome. Complicating the assessment of patients with connective tissue disease, PH may also occur in association with interstitial lung disease or left heart disease, or in isolation.
Patients with congenital heart disease, in whom the pulmonary circulation is exposed to increased blood flow and pressure from left-to-right shunts, may develop a pulmonary arteriopathy identical to that seen in other forms of PAH. If detected early, shunt closure may be feasible. In its most severe form, the shunt reverses, resulting in cyanosis and Eisenmenger syndrome, at which point shunt closure is contraindicated.
Pulmonary veno-occlusive disease (PVOD) and pulmonary capillary haemangiomatosis (PCH), now considered the same disease, are particularly rare and severe forms of PAH, in which progressive fibrosis of pulmonary venules occurs, resulting in severe hypoxaemia. Radiological clues seen on CT, such as septal thickening, centrilobular ground glass opacities and mediastinal adenopathy, can help identify patients with these forms of PAH, but a high index of suspicion is required. Standard PAH therapies may precipitate pulmonary oedema, and most patients with PVOD/PCH die or require lung transplantation within two years of diagnosis, making early referral crucial.7
Management
Management of patients with Group 1 PH consists of both specific PAH therapies, under specialist care, and the general measures detailed later in this article, which are handled in both primary care and specialist clinics. These treatments have been shown to drastically improve quality of life, exercise capacity and survival in this cohort. A small subset of patients will respond favourably to calcium channel blockers, indicated by a positive response to vasoreactivity testing during right heart catheterisation. For other patients, PAH-specific pulmonary vasodilator therapies (Table) are generally employed, usually in combination.
{kind=link}
Group 2. Pulmonary hypertension secondary to left heart disease
Accounting for nearly 70% of patients with PH in echocardiography-based studies,4 PH secondary to left heart disease (PH-LHD) most often results from heart failure with reduced ejection fraction, heart failure with preserved ejection fraction or valvular heart disease. Its exact prevalence is not known, partly due to variability in study designs and definitions used.
PH-LHD is thought to result initially from passive backward transmission of elevated left-sided filling pressures and loss of left atrial compliance, which then prompts a rise in pulmonary arterial pressures and reduced pulmonary vascular compliance. This is confirmed on right heart catheterisation by the presence of an elevated PAWP, reflecting raised left atrial pressure and indicating postcapillary PH. For some patients, this haemodynamic insult triggers pulmonary arterial endothelial dysfunction, decreased availability of vasodilatory compounds and consequent vascular remodelling.8 Patients in whom PH supervenes their cardiac disease often have more severe symptoms and exercise intolerance and markedly higher mortality.9
Management
The primary treatment for this group comprises diuretics, evidence-based therapies for the underlying heart failure or valvular disease, risk factor modification and treatment of comorbidities, such as obesity or atrial fibrillation. Studies into the use of PAH-specific therapies for patients with PH-LHD have been limited by the phenotypical diversity of this group, but they have thus far either failed to show a consistent benefit or have demonstrated harm.10,11 Unselective use of pulmonary vasodilators may precipitate hypotension, volume overload of the often noncompliant left heart system, and pulmonary oedema.
Group 3. Pulmonary hypertension secondary to chronic lung disease
Patients with advanced chronic lung disease (CLD), such as chronic obstructive pulmonary disease (COPD) or interstitial lung disease, often develop mild PH through a variety of pathophysiological mechanisms. Although this is common, the finding of severe PH in patients with CLD, particularly if radiological or physiological abnormalities are relatively mild, should prompt exclusion of superimposed insults, such as left heart disease or pulmonary emboli.
Conversely, patients with known CLD whose symptoms are disproportionately severe relative to their lung function test results should be further assessed with echocardiography. The development of PH in these patients is often heralded by worsening hypoxaemia and deteriorating functional capacity and carries a higher risk of dying.
Management
Treatment of this patient group is focused on the underlying disease rather than the PH itself. Long-term oxygen therapy is indicated for hypoxic patients and may reduce the rate of PH progression in patients with advanced COPD.12 Outside of carefully selected subsets of patients, PAH therapies generally cannot be recommended, as they may inhibit hypoxic vasoconstriction, thereby increasing blood flow to diseased lung with poor ventilation and worsening overall gas exchange.13
Group 4. Chronic thromboembolic pulmonary hypertension
The incidence of chronic thromboembolic PH (CTEPH) in survivors of acute pulmonary embolism is about 3%, with unprovoked and recurrent pulmonary embolism being significant risk factors. Occlusive thromboemboli that fail to resolve instigate a sustained rise in pulmonary vascular resistance, with nonoccluded areas developing a small vessel arteriopathy that is histologically indistinguishable from that of PAH.14
The combination of precapillary PH on right heart catheterisation and mismatched perfusion defects on ventilation–perfusion scanning after at least three months of effective anticoagulation treatment is indicative of CTEPH. Ventilation–perfusion scanning is the first-line imaging modality, with a sensitivity of up to 97% and specificity up to 95% for the detection of CTEPH.15 Invasive pulmonary angiography is the diagnostic gold standard, providing critical anatomical detail that also determines suitability for surgical or percutaneous interventions.
Management
Surgery is the treatment of choice for patients with disease in the proximal pulmonary arteries, and most patients who undergo pulmonary endarterectomy at an expert centre will experience significant symptomatic relief and near normalisation of their haemodynamics.16 Patients with inoperable disease, or those with persistent or recurrent PH after surgery, receive medical therapy with riociguat. Balloon pulmonary angioplasty, in which chronically stenotic pulmonary artery branches that are unsuitable for surgery are systematically dilated via endovascular techniques over multiple sessions, has seen a resurgence among patients in this group.
Lifelong anticoagulation therapy is recommended for all patients with CTEPH, regardless of treatment modality. These patients should all be assessed by an expert CTEPH team, as they may receive dramatic benefits from intervention if their disease is amenable.
Group 5. Pulmonary hypertension of unclear or multifactorial aetiologies
This varied group includes PH associated with a wide range of systemic, metabolic and haematological disorders, such as chronic haemolytic anaemia, sickle cell disease and myeloproliferative disorders. The mechanisms of PH in these diseases are poorly understood, and treatment of patients in this group focuses on the underlying diagnosis rather than the secondary PH. There is insufficient evidence for the use of PAH-specific therapies in this group.
General management of patients with pulmonary hypertension
In addition to the specified pharmacological measures for some groups, there are several recommended general measures for patients with PH, many of which will be handled in primary care.17 Monitoring of treatment adherence and recognition of medication side effects, pneumococcal and influenza vaccinations, correction of iron deficiency and diuretic therapy for patients with evidence of right heart failure are key aspects of management. The choice of diuretic varies by physician preference but, in all cases, attention to monitoring renal function and electrolyte levels and to avoiding excessive depletion of intravascular volume is required.
Atrial arrhythmias should be treated, with the goal of restoring sinus rhythm via cardioversion or ablation. Hypoxaemic patients with PaO2 less than 60 mmHg should receive supplemental oxygen. Management of comorbid medical conditions should be optimised. General anaesthesia is to be avoided where possible and carefully planned if required, with a preference for regional techniques.
Female patients should avoid pregnancy and use adequate birth control, owing to both the high risk of maternofetal mortality and the teratogenic potential of some PAH-specific medications, such as endothelin receptor antagonists. A combination of barrier and hormonal contraception methods is strongly recommended. As bosentan may reduce the efficacy of hormonal methods, a combination approach to contraception is essential for patients taking this drug.
Psychological support in the form of counselling or support groups is often welcomed by patients faced with a diagnosis of PAH, as many will not be familiar with the condition. Enrolment into a supervised exercise program tailored to disease severity, such as pulmonary rehabilitation, is also highly recommended.
When and where to refer patients
Most patients with suspected PH require specialist referral to a respiratory physician or cardiologist with expertise in PH assessment. Those suspected of having Group 1 (PAH) or Group 4 (CTEPH) disease, and those for whom pulmonary vasodilator therapy is being considered, should have a comprehensive assessment at an expert PH centre. A list of these centres in Australia is available on the Pulmonary Hypertension Association of Australia and New Zealand website (www.phsanz.org). These assessments typically comprise a clinical review, laboratory investigations to assess for complications or causative conditions, chest imaging, exercise testing, echocardiography and right heart catheterisation.
The therapeutic aim at an expert PH centre is for patients’ symptoms to stabilise or decrease and for patients to achieve stability or improvement in exercise capacity, quality of life and right ventricular function. These factors all indicate a low risk of dying as a result of PH. Higher risk patients whose symptoms are increasing, or who experience syncope or right ventricular failure, warrant early re-evaluation and escalation of therapy. Those who do not meaningfully respond to maximal medical therapy should be considered for a lung transplant assessment unless there are contraindications.18
Conclusion
PH encompasses a broad range of conditions. In its most frequently encountered forms, it simply represents a marker of severity for an underlying cardiac or respiratory disorder. It is the patients with the rarer forms of PAH or CTEPH who stand to benefit greatly from specific treatment, and identifying these patients early is crucial. Unfortunately, the symptoms of PAH are nonspecific, and delayed detection is common. By maintaining a high index of suspicion, along with knowledge of the trends in PH care and awareness of available expert PH centres, the GP has a key role to play in helping these patients access treatments that can greatly reduce their symptoms and improve their quality of life and prognosis. MT