It’s not a lipoma: when to suspect sarcoma and why specialised sarcoma centres matter

Professor Hong is Deputy Chair of the Australia and New Zealand Sarcoma Association; a Clinical Professor at the Central Clinical School, Faculty of Medicine and Health, The University of Sydney, Sydney; and a Radiation Oncologist at Chris O’Brien Lifehouse and Royal Price Alfred Hospital, Sydney, NSW.
Cancer
Soft tissue disorders
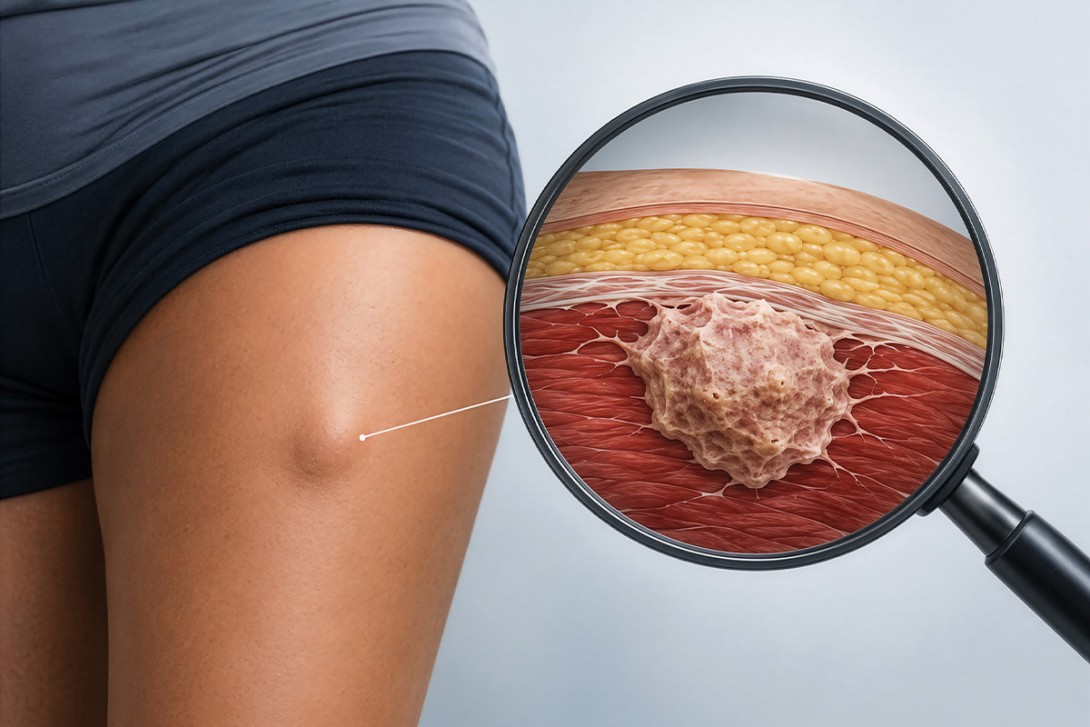
Sarcomas are rare, histologically diverse malignancies of bone and soft tissue. Every July, Sarcoma Awareness Month provides an opportunity to raise awareness of their wide spectrum of presentations and the importance of early specialist referral. This article outlines the clinical approach to suspected sarcoma, drawing on the recently published Australia and New Zealand Sarcoma Association clinical practice guidelines.
- Sarcomas encompass over 120 histological subtypes and may arise at any anatomical site, most commonly the extremities and retroperitoneum.
- Any soft tissue mass that is increasing in size, deeper than the fascia or larger than 5 cm warrants referral of the patient to a specialised sarcoma centre. Biopsy should not be performed before referral.
- Unplanned excision outside a specialised sarcoma centre significantly worsens local control, limb salvage rates and overall survival.
- The Australia and New Zealand Sarcoma Association guidelines recommend that all patients with suspected sarcoma be referred to a specialised sarcoma centre for multidisciplinary management.
- GPs play a pivotal role in early recognition, appropriate imaging and co-ordinating timely referral.
Sarcomas are rare malignancies that arise from mesenchymal tissues, encompassing bone and soft tissue. In 2024, there were about 2400 new diagnoses of soft tissue sarcoma and 276 new cases of bone sarcoma in Australia, representing about 1.5% of all new cancer diagnoses.1 Although rare in absolute terms, their clinical impact is disproportionate; sarcomas are the second leading cause of cancer-related death among Australians aged 0 to 19 years.1
For GPs, sarcomas present a diagnostic challenge. They may mimic common non-neoplastic conditions in otherwise well patients. They can arise at virtually any anatomical site and comprise 120 distinct histological subtypes with varied biological behaviours.2 Early recognition and prompt referral to a specialised sarcoma centre are among the most important determinants of outcome. A 2024 Australian Federal Parliament Senate inquiry into equitable access to rare cancer diagnosis and treatment specifically emphasised the need for accessible, GP-oriented clinical pathways for rare cancers including sarcoma.
This article provides a clinically oriented overview of sarcoma for GPs, covering presentations, the diagnostic approach and the latest evidence-based referral recommendations from the Australia and New Zealand Sarcoma Association (ANZSA) clinical practice guidelines.
Epidemiology and clinical importance
Soft tissue sarcomas account for about 1% of adult malignancies but about 15% of paediatric cancers.1 They affect individuals of all ages, with certain subtypes (e.g. rhabdomyosarcoma, Ewing sarcoma and osteosarcoma) predominating in children and young adults, whereas others (e.g. liposarcoma, leiomyosarcoma and undifferentiated pleomorphic sarcoma) are more common in older adults.
Bone sarcomas, although less frequent overall, have a similarly poor prognosis when diagnosed late. Osteosarcoma and Ewing sarcoma are the most common bone malignancies in the paediatric and young adult populations. Chordoma and chondrosarcoma are more often seen in adults. The rarity and heterogeneity of sarcomas mean that most treating clinicians outside specialised centres will encounter few cases in their career, making pattern recognition in general practice all the more important.
Histological diversity
The histological classification of sarcomas is complex and has evolved substantially with advances in molecular pathology.2 Over 120 distinct histological subtypes are recognised by the WHO. This heterogeneity is clinically important because the histological subtype determines prognosis and treatment planning (Table). For example, myxoid liposarcoma is highly radiosensitive, whereas synovial sarcoma responds to specific anthracycline-based chemotherapy regimens, and dedifferentiated liposarcoma has a different natural history from well-differentiated liposarcoma.3

Immunohistochemical and molecular characterisation are essential for accurate subtyping. Several sarcomas harbour pathognomonic translocations, such as the EWSR1–FLI1 fusion in Ewing sarcoma or the SS18–SSX fusion in synovial sarcoma. Targeted next-generation sequencing panels are increasingly being integrated into diagnostic workflows by specialised bone and soft tissue pathologists.4 This has direct therapeutic implications; for example, the identification of NTRK fusions allows for potential treatment with the targeted therapy larotrectinib.5
This complexity underscores why core needle biopsy and histopathological review must be performed within a specialised sarcoma centre with access to subspecialty pathology and molecular diagnostics.4
Clinical presentations: a wide spectrum
The range of clinical presentations is a defining feature of sarcomas and a source of diagnostic delay. There is no single pathognomonic symptom.
Soft tissue masses of the extremities
The most common presentation of soft tissue sarcoma is a palpable mass in the extremities, particularly in the thigh, shoulder girdle or upper arm. The mass is often painless in the early stages and may be mistaken for non-neoplastic causes, such as haematoma, or benign lesions, such as lipomas or ganglion cysts (Figures 1a, 1b and 1c). Features that should raise suspicion include:

- size greater than 5 cm
- deep-seated location
- increasing size over weeks to months
- firm or hard consistency.
The so-called ‘5 cm rule’ and depth criteria remain important clinical considerations, although smaller or superficial lesions can occasionally be malignant. Importantly, a palpable soft tissue mass should not be assumed to be benign and excised in a nonspecialised setting without appropriate cross-sectional imaging, such as CT with contrast or MRI, and specialist input.
Retroperitoneal and intra-abdominal presentations
Retroperitoneal sarcomas account for about 20% of soft tissue sarcomas and typically present late, as the retroperitoneum accommodates large tumours before symptoms emerge. Patients may report nonspecific abdominal fullness, early satiety, back pain or an incidentally detected abdominal mass (Figures 2a and 2b). Weight loss and lower limb oedema due to vascular or lymphatic compression can also occur. Liposarcoma and leiomyosarcoma are the dominant histological subtypes.

Bone sarcomas
Bone sarcomas most commonly present with localised pain, often initially attributed to musculoskeletal injury, growing pains in young patients or arthritis (Figure 3). A palpable mass may develop as the tumour expands through the cortex. Pathological fracture is an uncommon but serious presentation, particularly with lytic lesions.

Plain radiography often reveals abnormalities suggestive of bone sarcoma, with features such as cortical destruction, periosteal reaction (Codman’s triangle or ‘sunburst’ pattern) or a soft tissue mass. However, a normal x-ray does not exclude sarcoma, and cross-sectional imaging, such as CT with contrast and MRI, is required for full characterisation.
Other presentations
Uterine sarcomas (including leiomyosarcoma and endometrial stromal sarcoma) may present with abnormal uterine bleeding, pelvic pain or a rapidly enlarging uterus. These are often indistinguishable from benign fibroids on clinical assessment. Gastrointestinal stromal tumours may present with gastrointestinal bleeding and abdominal pain or may be found incidentally on imaging. Dermatofibrosarcoma protuberans presents as a slowly growing plaque or nodule on the trunk or proximal extremities, often initially misdiagnosed as a benign skin lesion or keloid (Figure 4).

The GP’s diagnostic approach
History and examination
For patients presenting with suspected sarcoma, the history should document:
- the duration
- the rate of growth
- any associated symptoms (e.g. pain, neurological symptoms, systemic features)
- any relevant past history
- any prior malignancy, including:
– prior radiotherapy
– family history of hereditary sarcoma syndromes, such as Li-Fraumeni syndrome, neurofibromatosis type 1 or hereditary retinoblastoma.
On examination, the size, depth, consistency, mobility, skin changes overlying the mass and regional lymph node status should be noted. Lymph node involvement is uncommon in most sarcoma subtypes, with the exception of rhabdomyosarcoma, epithelioid sarcoma and angiosarcoma. Vital signs and systemic examination should be performed to identify any features of metastatic disease.
Imaging
Plain radiography is the appropriate initial investigation for suspected bone lesions. For soft tissue masses, MRI of the primary site is the investigation of choice. MRI can help define anatomical relationships, tissue characteristics and neurovascular involvement, which are critical for surgical planning. Whole-body 18 F-fluorodeoxyglucose positron emission tomography/CT and chest CT are required for systemic staging, as sarcomas most commonly metastasise to the lungs.
What not to do
A key point for general practice: do not biopsy a suspected sarcoma without prior input from a specialised sarcoma team. An unplanned excision or incorrectly performed biopsy can compromise the subsequent surgical field, contaminate tissue planes and significantly worsen local recurrence rates and limb salvage options.
Similarly, the temptation to aspirate a fluctuant mass or excise a ‘lipoma’ with suspicious features – such as rapid growth over weeks, pain, firmness and a deep-seated location – must be resisted (Box). If in doubt, arrange an MRI and refer the patient to a sarcoma specialist.

The contact details of specialised sarcoma centres in Australia and New Zealand are available on the ANZSA website (https://www.sarcoma.org.au). Most centres have a clinical nurse consultant or co-ordinator who can facilitate urgent triage and advise on appropriate workup before the first specialist appointment.
Clinical practice guidelines: evidence for specialised sarcoma care
The ANZSA commenced guideline development in 2020, publishing evidence-based recommendations on three priority topics:
- the role of specialised sarcoma centres (incorporating surgery and radiotherapy)
- management of retroperitoneal sarcoma
- management of paediatric, adolescent and young adult patients with sarcoma.6
These guidelines are freely available online (at https://www.sarcoma.org.au) and include plain-language summaries in multiple languages for patients.
Does surgery at a specialised sarcoma centre improve outcomes?
A systematic review of 66 retrospective studies, many from large national cancer registries, demonstrated consistent evidence that surgery at specialised sarcoma centres is associated with improved outcomes across multiple endpoints.7 Six studies, including one with over 14,000 patients from the USA, found that treatment at high-volume centres was associated with a significantly increased likelihood of limb-salvage surgery compared with amputation in primary bone tumours (odds ratio, 1.34; 95% confidence interval, 1.14–1.59; p = 0.001).7 Seven of 12 studies reporting perioperative mortality found lower rates at high-volume centres.7 Sixteen of 21 studies assessing overall survival demonstrated that surgery at a specialised centre was an independent predictor of improved survival.7
The ANZSA guidelines working party issued a Grade B recommendation that patients with suspected sarcoma be referred to a specialised sarcoma centre for management to reduce local recurrence, minimise surgical complications, improve limb conservation and improve survival.8
Does radiotherapy at a specialised sarcoma centre improve outcomes?
A systematic review of 21 studies examined the effect of radiotherapy at specialised versus nonspecialised centres.9 The key importance of radiotherapy plan quality was highlighted by the Preoperative radiotherapy plus Surgery versus Surgery alone for patients with primary Retroperitoneal Sarcoma (STRASS) trial of preoperative radiotherapy for retroperitoneal sarcoma, in which patients treated with a noncompliant radiotherapy plan had significantly worse abdominal relapse-free survival compared with those with a compliant plan.10 Two studies in the systematic review demonstrated a correlation between centre volume, use of radiotherapy and local disease control.9
The ANZSA recommends (Grade B) that patients with soft tissue sarcoma requiring radiotherapy be managed through a specialised sarcoma centre to reduce local recurrence and major wound complications.8
Multidisciplinary team care: the gold standard
The management of sarcoma is fundamentally multidisciplinary, requiring close collaboration between surgical oncology, orthopaedic surgery, radiation oncology, medical oncology, subspecialty pathology (including molecular diagnostics), radiology, physiotherapy, allied health and palliative care.
Multidisciplinary review enables:
- accurate histological subtyping with subspecialty pathology input and correlation with radiology
- appropriate staging and preoperative imaging interpretation
- planning of limb-conserving or function-preserving surgery where possible
- radiotherapy planning, including decisions regarding preoperative
- versus postoperative sequencing and dose prescription
- consideration of neoadjuvant or adjuvant systemic therapy, assessment and monitoring of treatment-related toxicity, including cardiac function
- access to clinical trials, which are often available only at specialised sarcoma centres
- multidisciplinary rehabilitation planning for functional recovery.
Special populations
Children, adolescents and young adults
Sarcomas in children and adolescents require paediatric oncology expertise and are managed via age-specific protocols. Multidisciplinary co-ordination between adult and paediatric sarcoma services is increasingly recognised for adolescents and young adults, who may be managed across both settings depending on age cutoffs and institutional expertise.
Rural and remote patients
Access to specialised sarcoma care represents a significant equity challenge for patients residing in rural and remote Australia. Telehealth multidisciplinary team input, co-ordinated care models and referral networks with major centres have improved access, but geographical barriers remain. GPs in these regions play an especially important role in early recognition and advocacy for timely referral. Travel support programs and patient accommodation assistance are available through many cancer centres and state-based services.
Patients with hereditary cancer syndromes
Certain hereditary syndromes confer an elevated sarcoma risk. Li-Fraumeni syndrome (TP53 germline mutation) predisposes individuals to soft tissue sarcoma, osteosarcoma and other malignancies. Neurofibromatosis type 1, caused by germline mutations in NF1, is associated with malignant peripheral nerve sheath tumours, a highly aggressive sarcoma with a poor prognosis. Hereditary retinoblastoma survivors are at significant risk of subsequent sarcoma, particularly after radiotherapy. Any sarcoma diagnosis in a young patient, or a patient with a relevant personal or family history, warrants referral for genetic counselling.
The GP’s ongoing role in sarcoma care
For GPs, the decision to refer a patient to a specialised sarcoma centre should not be deferred while awaiting further investigations or a histological diagnosis. Referral should occur on the basis of clinical and imaging suspicion. Biopsy, histological subtyping and staging are best co-ordinated within the multidisciplinary framework of a specialised sarcoma centre.
The GP’s role does not end at referral. In the management of sarcoma, GPs are pivotal in:
- co-ordinating surveillance imaging, as recommended by the treating sarcoma team
- monitoring for late effects of treatment, including lymphoedema, neuropathy, joint stiffness and cardiotoxicity in patients who have received anthracyclines
- providing psychological support, as a diagnosis of sarcoma carries a significant psychological burden, particularly in younger patients
- recognising symptoms potentially indicative of relapse, including new pulmonary symptoms (the lungs being the most common site of metastatic disease), local pain or new masses
- supporting patients on treatment with medication management, monitoring for drug interactions and liaising with the specialist team
- advance care planning in patients with advanced sarcoma
- support for other family members.
Conclusion
Sarcomas are rare and aggressive malignancies that span a wide spectrum of histological subtypes, anatomical sites and patient demographics. Although individual cases are uncommon in general practice, awareness of the warning features of a suspicious soft tissue or bone mass, a low threshold for MRI and timely referral to a specialised sarcoma centre are critical.
The ANZSA clinical practice guidelines provide clear, evidence-based guidance: patients with suspected sarcoma should be referred promptly to a specialised sarcoma centre for multidisciplinary assessment, biopsy and management planning. The benefit of specialised care extends across surgical outcomes, limb salvage, radiotherapy outcomes and overall survival. MT
COMPETING INTERESTS: Professor Hong has received advisory board payments from Oncobeta and Telix.
References
1. Australian Institute of Health and Welfare (AIHW). Overview of cancer in Australia, 2025. Canberra: AIHW; 2025. Available online at: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/contents/overview#rare (accessed June 2026).
2. WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. WHO Classification of Tumours, 5th ed., vol. 3. Geneva: WHO; 2020.
3. Lansu J, Bovee J, Braam P, et al. Dose reduction of preoperative radiotherapy in myxoid liposarcoma: a nonrandomized controlled trial. JAMA Oncol 2021; 7: e205865.
4. Vu J, Petrucco C, Vargas C, et al. The value of a second expert opinion in histopathological diagnosis of bone and soft tissue sarcoma: a systematic review. Pathology 2026; 58(1): 1-7.
5. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020; 21: 531-540.
6. Gyorki DE, Bae S, Smith RC, et al. Update of clinical practice guidelines for the management of patients with sarcoma. ANZ J Surg 2025; 95: 289-292.
7. Hong AM, Sundaram A, Perianayagam G, et al. Surgery at specialised sarcoma centres improves patient outcomes - a systematic review by the ANZSA clinical practice guidelines working party. Eur J Surg Oncol 2023; 49: 106951.
8. Australia and New Zealand Sarcoma Association (ANZSA). Clinical practice guidelines for management of sarcoma. Executive summary. Melbourne: ANZSA; 2026. Available online at: https://sarcoma.org.au/pages/sarcoma-guidelines/executive-summary (accessed June 2026).
9. Hong AM, Lo H, Lawless A, et al. The benefit for RT at specialised sarcoma centres: a systematic review and clinical practice guidelines from ANZSA. Radiother Oncol 2022; 177: 158-162.
10. Haas R, Stelmes JJ, Zaffaroni F, et al. Critical impact of radiotherapy protocol compliance and quality in the treatment of retroperitoneal sarcomas: results from the EORTC 62092-22092 STRASS trial. Cancer 2022; 128: 2796-2805.