Systemic sclerosis. A complex multisystem disease

Systemic sclerosis is a rare disease characterised by vascular abnormalities and fibrosis of multiple organs. Proactive screening and follow up for organ-specific complications are important aspects of management.
Systemic sclerosis (SSc), also known as scleroderma, is a rare autoimmune condition with high morbidity and mortality caused by multisystem organ involvement secondary to vasculopathy, fibrosis and immune system alteration.1 SSc is more common in women than men and has an estimated prevalence of 20 per 100,000 population in Australia.1 Over the last few decades, however, there has been an improvement in outcomes and mortality, which has been attributed to better recognition of the disease and proactive screening for organ-specific complications.2,3 This article will discuss the common manifestations of SSc and provide guidance about shared care with rheumatologists for patients with this condition.
How do patients with systemic sclerosis present?
There is significant clinical variability in patient presentations of SSc, and clinicians should be aware of distinct manifestations and complications for early disease compared with late disease.4 Patients can be classified according to the extent of their skin thickening or hardening with:
- limited cutaneous SSc (LcSSc), or
- diffuse cutaneous SSc (DcSSc).
There is a predominance of the limited subtype, which accounts for 70 to 80% of SSc presentations.1
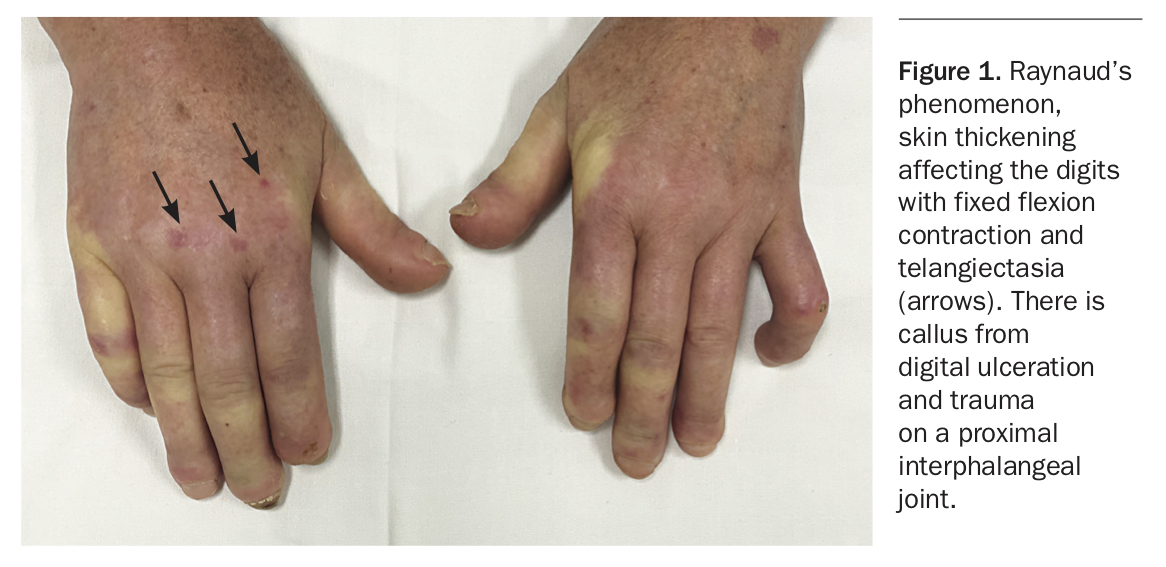
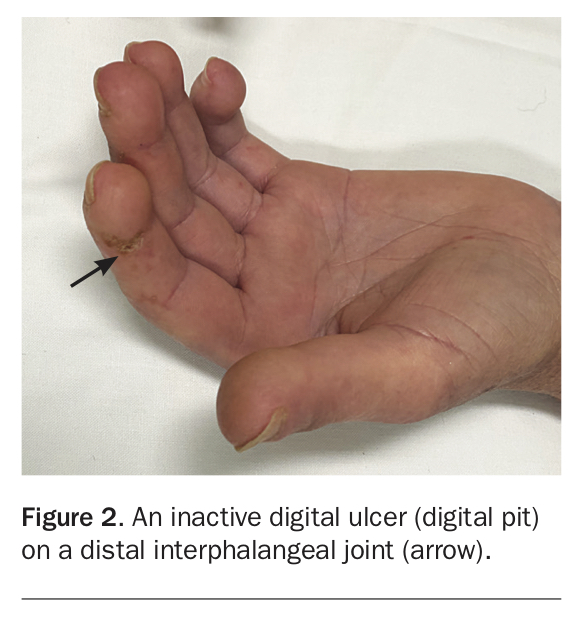
SSc is characterised by vascular abnormalities and fibrosis of multiple organs; a summary of common manifestations is presented in Box 1.4-6 Although there are clinical characteristics that are shared in both LcSSc and DcSSc, some features are associated with one subtype (Box 2). Patients with either LcSSc or DcSSc may have Raynaud’s phenomenon, which occurs in 95% of all patients (Figure 1) – in patients with LcSSc this may have been present for many years before the development of other features. Ischaemic digital tip ulcers and chilblains are complications of Raynaud’s phenomenon and are prone to developing infections as a result of poor wound healing (Figure 1 and Figure 2).4 Patients may have small pitting scars on the digital tips which are evidence of previous digital ulcers that have healed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
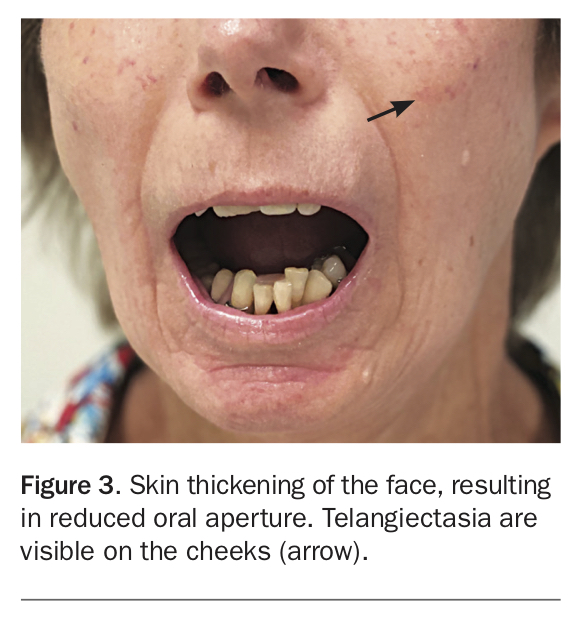
The complications of early skin thickening or fibrosis may become apparent at any stage of the disease and may involve the hands or face (Figure 1, Figure 2 and Figure 3). Patients may develop joint contractures of the digits, with inability to flex or extend the digits or wrists causing significant functional impairment. This is more likely in patients with DcSSc and has been associated with anti-topoisomerase antibody (anti-Scl70 antibody).5 Skin and musculoskeletal manifestations may be more pronounced in the early phases of DcSSc. Patients may present with generalised ‘puffiness’ of the fingers and hands, or with an inflammatory arthritis, myalgia or even mild myositis – these manifestations can occur in LcSSc but are often more florid and extensive in DcSSc.4
{kind=link}
Patients with either LcSSc or DcSSc frequently have gastrointestinal involvement, which may be due to vascular or fibrotic complications. The most common gastrointestinal manifestation is gastro-oesophageal reflux disease, which can be present in 80% of all patients with SSc.3 Patients may also present with iron deficiency anaemia secondary to upper gastrointestinal bleeding caused by gastric antral vascular ectasia. They may also have complications of gastrointestinal dysmotility due to fibrosis, such as small intestinal bacterial overgrowth or faecal incontinence.6
Up to 80% of all patients with SSc have interstitial lung disease (ILD), with most patients with ILD developing restrictive lung disease in the first five years after the onset of symptoms. Progressive lung disease occurs in 25 to 30% of patients.4
One of the major contributors to mortality in patients with SSc is the development of pulmonary arterial hypertension (PAH), which can occur at any time in patients with either LcSSc or DcSSc. Up to 10% of patients are affected by PAH.1
When can a diagnosis of systemic sclerosis be made?
A diagnosis of SSc can be considered when a patient meets the classification criteria for SSc defined by the American College of Rheumatology and the European League Against Rheumatism (ACR/EULAR) in 2013.7 These criteria include a combination of clinical features and positive blood tests for SSc-associated autoantibodies: anticentromere antibody, anti-Scl70 antibody and/or anti-RNA polymerase III antibody.7 Classification criteria have been developed because of the hetergeneous presentation of SSc, primarily for use in clinical research to ensure a homogeneous selection of patients; however, these criteria can also serve to support a diagnosis of SSc.
A diagnosis of SSc should also be considered if a patient presents with Raynaud’s phenomenon and additional clinical features (see Box 1) and has a positive antinuclear antibody (ANA) or positive anti-extractable nuclear antibody test result.
What causes systemic sclerosis?
Although the aetiology and pathogenesis of SSc remain poorly understood, epidemiological studies have shown strong evidence of familial clustering of cases, and increased risk in first-degree relatives of patients with SSc.8 It is thought that susceptible individuals develop the disease after stimulation by initiating events.8 There have been associations with occupational exposures, particularly exposure to vinyl chloride; more recently, development of a scleroderma-like disease has been observed in stonemasons exposed to dust from artificial stone.8,9
What are the complications of systemic sclerosis?
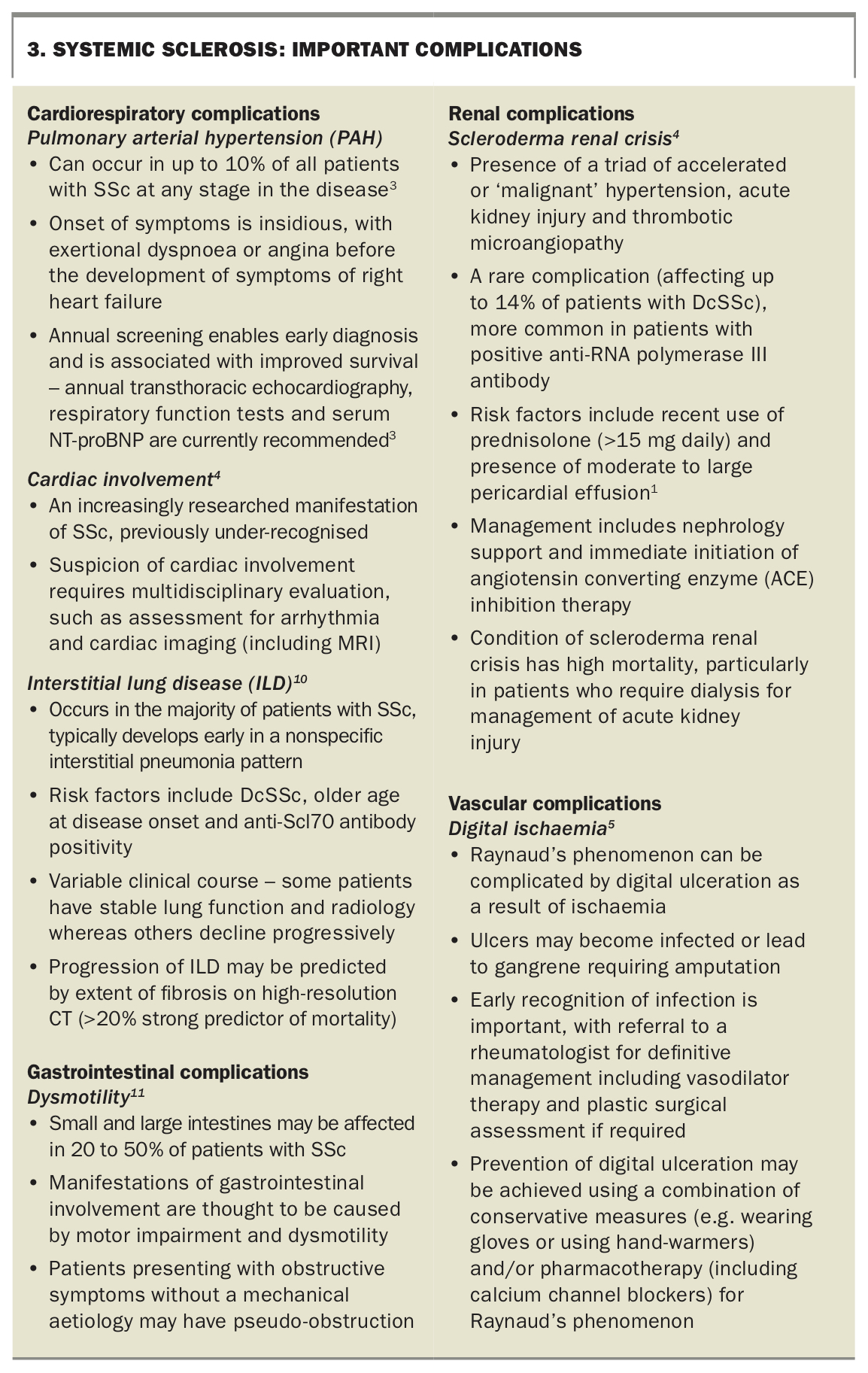
There are multiple complications of SSc that should not be missed. A summary of the important potential complications is provided in Box 3.1,3-5,10,11
{kind=link}
What investigations should be ordered?
Initial investigations should aim to confirm a diagnosis of SSc and evaluate the presence of organ-specific complications.4
If SSc is suspected, blood tests for autoantibodies are required to confirm the diagnosis and help to predict the disease subtype, as patients with LcSSc have a better prognosis.12 ANA is present in almost all patients with SSc (93 to 96%). Anticentromere antibody is associated with LcSSc whereas anti-Scl70 is associated with DcSSc.1
Baseline evaluation of organ involvement should include transthoracic echocardiography and respiratory function tests (RFTs) to investigate for ILD and PAH. In the setting of results from RFTs demonstrating reduced forced vital capacity (FVC) and/or reduced diffusing capacity for carbon monoxide (DLCO), the presence of ILD should be further investigated with high-resolution CT of the chest. It should be noted, however, that a DLCO less than 70% with a relatively preserved FVC is associated with a diagnosis of PAH.13,14 A thorough history and examination for symptoms and signs of gastrointestinal involvement should guide symptom-based investigations, such as iron studies or endoscopy, as required.
For patients with anti-RNA polymerase III antibody positivity or early DcSSc, blood pressure monitoring is recommended three times per week, with assessment of renal function and urinalysis at regular intervals to screen for scleroderma renal crisis.4 International guidelines recommend annual transthoracic echocardiography, RFTs and serum NT-proBNP to screen for the development of ILD and PAH in all patients with SSc.13
What does shared care involve?
In recent decades, outcomes and survival for patients with SSc have improved, which has been attributed to improved recognition of organ-specific complications and proactive screening and follow-up.4 Any patient with suspected SSc should be referred to a rheumatologist or specialised centre with appropriate expertise in SSc management for confirmation of the diagnosis and advice regarding organ-specific complications to be aware of during community follow-up.15 Patients who develop progressive lung disease require the input of respiratory physicians or rheumatologists to guide use of immunosuppressive therapies.4
The approach to treatment of SSc is tailored to an individual patient’s phenotype and based on international guidelines.12 Involvement of other allied health specialists, including occupational therapists, physiotherapists, dietitians and speech pathologists, can improve care of patients with SSc.15
What about follow up and prevention?
Important aspects of management for patients with SSc in primary care are discussed in Box 4.16
{kind=link}
Patients with SSc also have a higher prevalence of cancer, and age-appropriate malignancy screening is recommended as part of routine community follow up.1
Conclusion
It is important that all clinicians are aware of the common manifestations of SSc, which are often first seen in general practice. Regular communication and shared care with a rheumatologist will provide guidance for the risk, presence and severity of organ-specific complications relevant in the care of patients with SSc. MT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.