Childhood-onset systemic lupus erythematosus: a clinical update
Dr Power is a Paediatric Rheumatology Fellow in the Department of Paediatric Rheumatology, Monash Children’s Hospital, Melbourne.
Dr Renton is a Paediatric Rheumatologist in the Department of Paediatric Rheumatology, Monash Children’s Hospital, and the Rheumatology Team, Department of General Medicine, The Royal Children’s Hospital, Melbourne.
Dr Tiller is a Paediatric Rheumatologist and General Paediatrician in the Department of Paediatric Rheumatology, Monash Children’s Hospital, and the Rheumatology Team, Department of General Medicine, The Royal Children’s Hospital, Melbourne, Vic.
SERIES EDITOR: Associate Professor Arvin Damodaran BSc, MB BS(Hons), MMedEd, FRACP, representing the Education Training and Workforce Committee of the Australian Rheumatology Association.

Lupus
Connective tissue disorders

Systemic lupus erythematosus (SLE) is the most common autoimmune connective tissue disorder of childhood. This clinically heterogeneous disease has a more aggressive phenotype in children than in adults. Prompt recognition of the key features of childhood-onset SLE by a general practitioner enables early diagnosis and treatment by a paediatric rheumatologist.
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder involving systemic inflammation that affects multiple organs. Recognising the signs and flares of SLE in children in general practice is important, followed by its diagnosis and treatment by a rheumatologist. This article describes the stepwise approaches to the diagnosis and management of childhood-onset SLE (cSLE).
Epidemiology
SLE is a complex, multisystem disease of immune dysregulation, with alterations in both innate and adaptive immunity.1 This chronic, heterogeneous disease presents with a variety of clinical manifestations including cutaneous, musculoskeletal, haematological, renal and neurological involvement. SLE carries a significant burden of disease, with high morbidity and mortality rates.2 The estimated incidence and prevalence of SLE vary considerably among geographical locations.3 Australia-specific data on SLE are limited, with figures suggesting a prevalence ranging from 19 per 100,000 in people of European ancestry to 92 per 100,000 in Indigenous Australians.4 There are higher rates of SLE in non-white populations including Indigenous, Latino, Asian and
Afro-Caribbean populations.5
Patients with cSLE account for an estimated 15 to 20% of all patients with SLE.6,7 The average age of onset of cSLE is 12 years, with rare cases occurring in children younger than 5 years of age.8 The first longitudinal study of Australian children with SLE found Indigenous Australians made up a high proportion (26%) of the cohort of 42 children, despite only making up 3.1% of the Western Australian population.9
Differences in the clinical phenotype and serological profile of cSLE versus adult-onset SLE are well described in the literature.10–12 Patients with cSLE typically have more aggressive disease than their adult counterparts, with higher reported frequencies of lupus nephritis, central nervous system involvement and haemolytic anaemia when compared with cohorts of adults with lupus.13,14 The sex distribution of cSLE is about five to one female to male, compared with 10 to one in the adult population.15
Key clinical manifestations of childhood-onset SLE
The presenting symptoms of cSLE may be subtle. However, most patients have severe disease. The disease may initially present in a single organ but most commonly manifests as a multiorgan disease. The characteristic features are listed below.
- Constitutional symptoms:
– fever, generalised malaise, weight loss, loss of appetite, fatigue. - Mucocutaneous features:
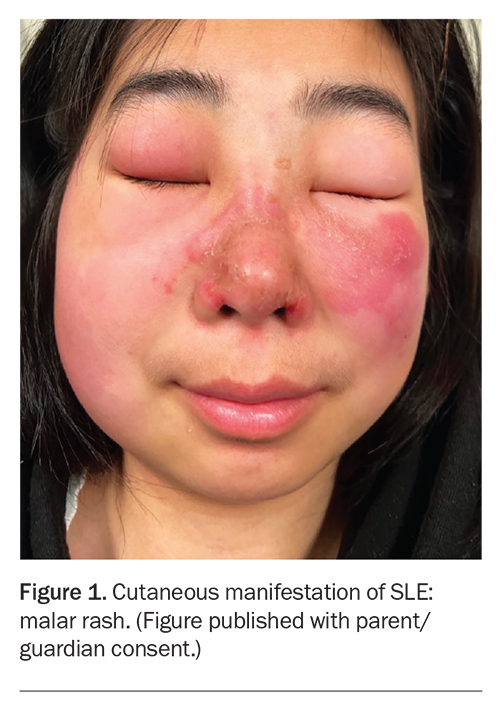
– a malar or ‘butterfly’ rash – a fixed erythematous rash that may be flat or raised and characteristically occurs over the malar eminences, sparing the nasolabial folds (Figure)
– a discoid rash – raised erythematous patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions
– photosensitivity – a skin rash that occurs as a result of an unusual reaction to sunlight
– oral or nasal ulcers, or both – usually painless; hard palate erythema is often observed
– alopecia – occurs without scarring; tends to occur at the frontotemporal hairline but can be more diffuse
– subungual erythema
– other cutaneous findings, including a maculopapular rash, bullous lupus, a toxic epidermal necrolysis variant of lupus, a psoriasiform rash, an annular rash, verrucous lupus, lupus erythematosus tumidus, chilblain lupus and discoid lupus/lichen planus overlap. - Musculoskeletal features:
– arthralgia and myalgia are common initial symptoms
– arthritis may be a prominent feature in some patients, with typical joint swelling, loss of range of motion, tenderness, and pain on motion. - Renal features:
– glomerulonephritis, nephrotic syndrome, hypertension
– kidney disease is a significant cause of morbidity and mortality in paediatric patients
– about 20 to 75% of children with SLE will develop nephritis, with 18 to 50% progressing to end-stage kidney disease. - Serositis:
– pericardial effusion or pericarditis; pleural effusion. - Neuropsychiatric features:
– acute confusional state, seizures, psychosis, cognitive defects, chorea, cerebrovascular accidents, pseudotumour cerebri. - Gastrointestinal features:
– lupus mesenteric vasculitis (lupus enteritis), mesenteric thrombosis, hepatic thrombosis, pancreatitis, acalculous cholecystitis, lupus-related protein-losing enteropathy, pseudo-obstruction, ascites, peritonitis, deranged liver function test results. - Reticuloendothelial features:
– diffuse lymphadenopathy, hepatosplenomegaly, Kikuchi lymphadenitis.
{kind=link}
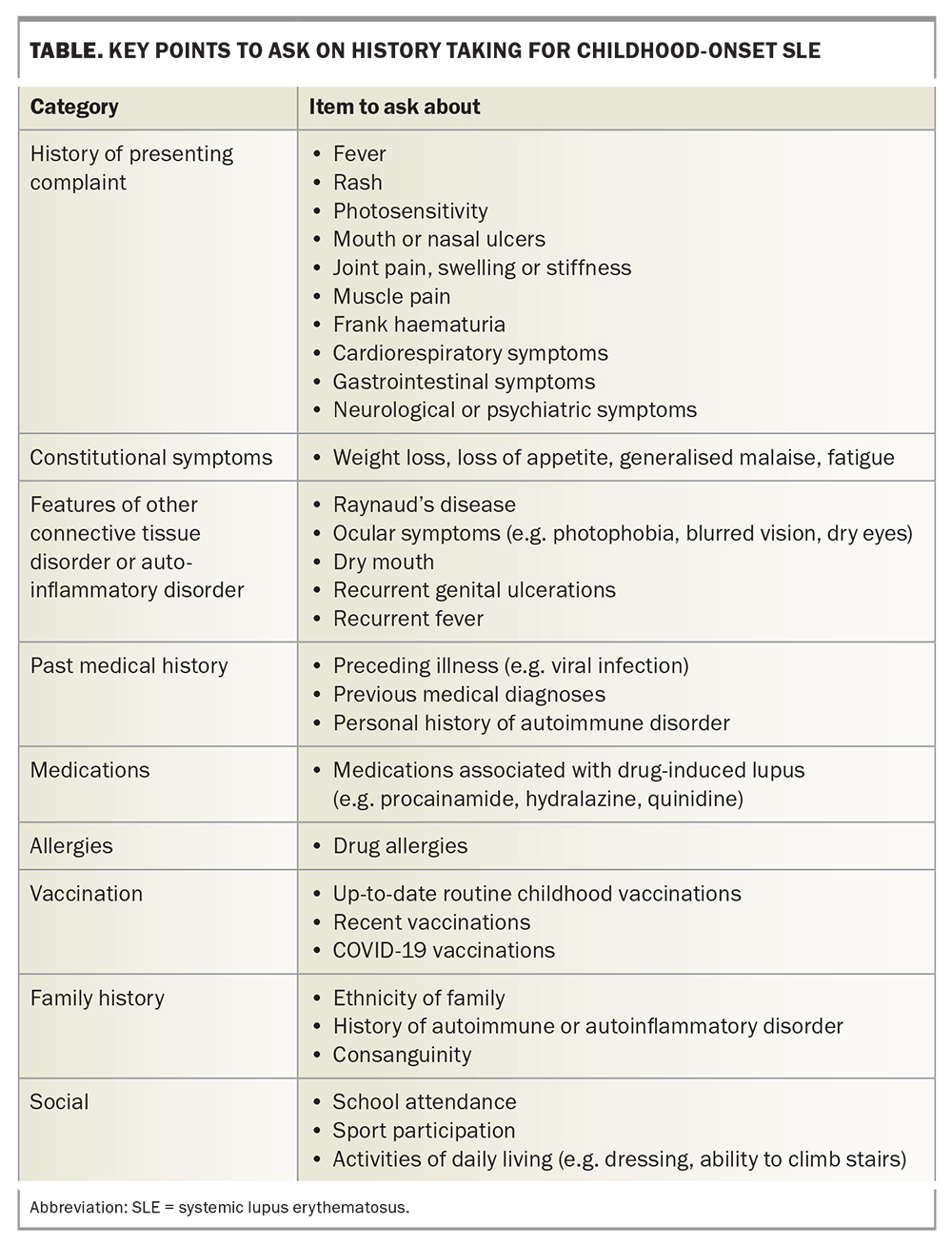
History taking and clinical examination
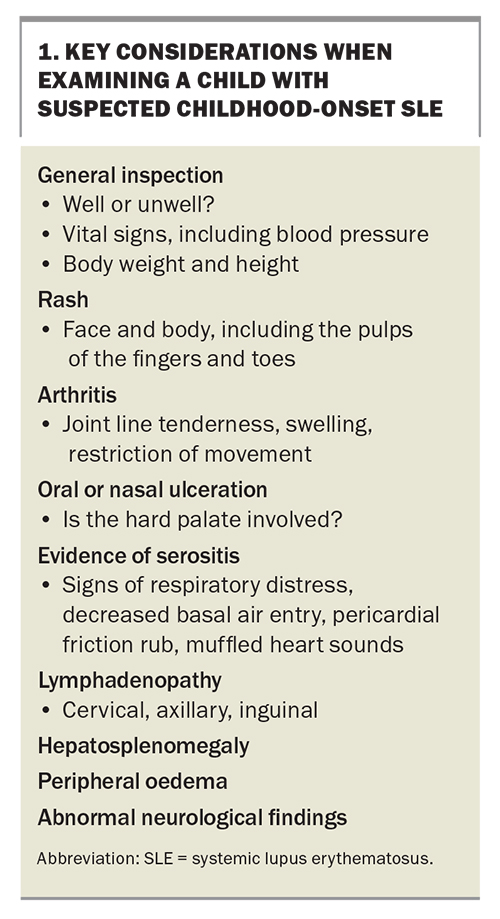
A thorough history and examination are required for all patients with suspected SLE. The signs and symptoms to ask about are listed in the Table, and the features to consider on examination are listed in Box 1.
{kind=link}
{kind=link}
Investigations for cSLE
The investigation of a child with suspected SLE in general practice should begin with measurements of body weight, height and blood pressure. The following investigations are then performed:
- full blood examination, including reticulocytes
- blood film test
- assessments of urea, electrolytes and creatinine levels
- liver function tests
- coagulation test
- assessments of erythrocyte sedimentation rate (ESR) and C-reactive protein levels (CRP) levels
- antinuclear antibody (ANA) test
- urine dipstick test
- urine microscopy, culture and sensitivity test
- measurement of urine protein:creatinine ratio.
It may be prudent to obtain an additional serum sample to avoid repeated blood sampling in younger children.
Antibody tests for SLE
ANA testing has an important role in identifying autoimmune connective tissue disorders, including SLE. However, this test has limitations. Although it has a high negative predictive value for excluding connective tissue disorders as a cause for a patient’s symptoms, in isolation, a positive ANA result has a low positive predictive value for diagnosing connective tissue disorders.16 It is a nonspecific test, with a reported frequency of a positive result of between 0 and 70% in healthy children depending on the testing methodology.17 ANA positivity can also be associated with infection (influenza A, Epstein-Barr virus, tuberculosis, HIV, subacute bacterial endocarditis) and malignancy (non-Hodgkin’s lymphoma, epithelial ovarian carcinoma).18-21 Therefore, the authors strongly recommend against routine ANA testing when a clinician has a low suspicion of a diagnosis of SLE in a patient. Likewise, serial ANA testing after a negative result has low clinical utility and is costly.22
A stepwise approach to antibody testing is generally recommended for the diagnosis of SLE. In the context of a positive ANA test result and strong clinical suspicion for SLE, further investigation with an anti-double-stranded DNA (anti-dsDNA) test, an extractable nuclear antigen (ENA) test and complement C3 and C4 tests are appropriate (Box 2). Anti-dsDNA antibodies are present in 72 to 93% of children with SLE, making them a highly sensitive marker. The reported specificity of anti-dsDNA is 97%.23 An ENA panel detects a broad set of autoantibodies associated with connective tissue disorders, including
anti-Smith, anti-Ro/La, antinuclear ribonucleoprotein (anti-RNP), anti-Jo-1 and anti-Scl70. Although anti-Smith antibodies have low sensitivity for SLE, they are highly specific, with a reported specificity of 99%.24
{kind=link}
Diagnosis
Important laboratory findings in patients with cSLE include:
- haemolytic anaemia (reduced haemoglobin, raised reticulocyte count, reduced haptoglobins, direct Coombs test [DCT] positivity, raised indirect bilirubin)
- leukopenia
- lymphopenia
- thrombocytopenia
- renal dysfunction (raised urea and creatinine levels, reduced estimated glomerular filtration rate (GFR), microscopic haematuria, proteinuria, casts, raised white and red cell counts in urine when infection is excluded)
- raised transaminase levels
- elevated levels of inflammatory markers (ESR, CRP)
- hypocomplementaemia (reduced C3 and C4 levels)
- immunological findings:
- ANA positivity
- antiphospholipid antibodies (anticardiolipin antibody, beta-2 glycoprotein 1, lupus anticoagulant)
- SLE-specific antibodies (anti-dsDNA, anti-Smith antibodies).
Classification criteria
Several classification criteria have been developed to assist clinicians with the diagnosis of SLE and to homogenise study cohorts for research purposes. The American College of Rheumatology (ACR) developed a classification system in 1971, which was most recently modified in 1997.25,26 According to these revised criteria, four of 11 ACR criteria must be found in a patient to fulfil the classification of SLE. These criteria include a malar rash, a discoid rash, photosensitivity, oral and nasal ulcerations, serositis, ANA positivity, and specific renal, neurological, haematological and immunological disorders.
The Systemic Lupus International Collaborating Clinics (SLICC) published a new set of criteria in 2012.27 These criteria involve 11 clinical and six immunological items, requiring four items with at least one clinical and one immunological item, or biopsy-proven nephritis compatible with lupus in the presence of ANA or anti-dsDNA positivity. The SLICC criteria have improved sensitivity with slightly decreased specificity compared with the ACR criteria.28 The European League Against Rheumatism/ACR classification criteria require the presence of ANAs at a titre of 1:80 or greater as an ‘entry criterion’.29 Clinical and laboratory criteria are weighted, with a total score of 10 or more meeting the requirements for the classification of SLE.
Although these criteria are undoubtedly valuable, physicians should use clinical judgement to determine the diagnosis in patients on an individual basis.
Management
General management
Early referral to a paediatric rheumatologist is advised for all patients with suspected SLE. General practitioners are a crucial liaison between the hospital and community services for patients with SLE and are often the first medical point of contact to diagnose disease flares, comorbidities, medication toxicities and disease-related damage. Management of this chronic disease requires effective communication between the general practitioner and specialist service in order to provide high-quality patient- and family-centred care. Other vital members of the multidisciplinary team include the clinical nurse specialist, physiotherapist, occupational therapist, psychologist, social worker and dietitian.
The aims of treatment are to control disease activity, prevent permanent organ damage, minimise medication toxicity and optimise a patient’s quality of life. Providing education and support to patients and their families through a multidisciplinary approach is fundamental to achieving disease control. General lifestyle advice includes following a healthy diet and exercise regimen, sleep hygiene, stress management, daily application of high-protection (sun protection factor 50+) sun cream and wearing of a wide-brimmed hat and long-sleeved clothing.
Immunisation
Immune dysregulation and immunosuppressive treatments lead to a markedly increased risk of infections in patients. Therefore, routine childhood vaccinations, annual influenza vaccinations and additional vaccines (including pneumococcal and meningococcal) are strongly advised for high-risk populations. COVID-19 vaccination as per state and national guidance is also recommended for immunosuppressed patients.
Recommendations regarding live vaccinations in immunosuppressed patients should be considered on an individual patient basis. The Australian Immunisation Handbook suggests that live vaccines are contraindicated for patients who are on high-dose immunosuppressive therapies.30 These guidelines propose that patients who are mildly immunocompromised (patients receiving selected conventional, synthetic disease-modifying antirheumatic medications [DMARDs] in low doses, either on their own or in combination with low-dose corticosteroids) may be eligible for some live vaccines but recommend discussion with a specialist to carry out an individualised risk assessment.
In practice, live vaccines are generally avoided in all patients on any DMARD treatment. If there is uncertainty about the safety of vaccination in individual circumstances, expert advice from their treating physician or an immunisation specialist should be sought prior to vaccination.
Pharmacological therapy
The choice of pharmacological agent is individualised, primarily based on the patient’s disease manifestations, severity of disease, tolerability of side effects and patient or family preference.
Corticosteroids and NSAIDs
Corticosteroids are the cornerstone of the initial management of cSLE. Patients may require high-dose oral or intravenous corticosteroids at diagnosis, with a tapering regimen provided thereafter, depending on the subsequent clinical course. ‘Sick-day management’ with higher corticosteroid doses should be considered for patients on long-term corticosteroid treatment and discussed with the treating team. Corticosteroids should not be abruptly discontinued in those who have been taking them long-term because of the risk of adrenal insufficiency.
Paediatric patients may have significant morbidity from the prolonged use of high-dose corticosteroid therapy. Potential side effects include weight gain, short stature, striae, cataracts and glaucoma, osteoporosis, impaired glucose tolerance, hypertension, sleep disturbance and behavioural issues.
NSAIDs (ibuprofen, naproxen, meloxicam, celecoxib) are useful adjuncts to treatment, particularly for musculoskeletal symptoms. Patients with renal insufficiency should avoid this class of medication.
DMARDs
The choice of DMARD should be guided by specific organ involvement and under the care of a specialist with expertise in cSLE.31 Hydroxychloroquine is a widely used medication in lupus management, leading to improvements in rash, alopecia and musculoskeletal symptoms. It is also reported to play a role in reducing disease flares.32 Hydroxychloroquine is commonly used as a maintenance therapy because of its low toxicity, and monotherapy with hydroxychloroquine may be sufficient for children with mild disease. Of note, retinal toxicity after five years of therapy becomes a concern, and patients require ophthalmological monitoring.
Methotrexate is often used as a corticosteroid-sparing agent for mild to moderate disease. It has shown efficacy in skin, joint and mucocutaneous involvement. Mycophenolate and azathioprine are also often used for lupus. Genotyping of thiopurine methyltransferase, which metabolises azathioprine, may help identify those at risk of toxicity. Cyclophosphamide is typically reserved for severe SLE, especially that with kidney, central nervous system and other severe organ involvement. The use of cyclophosphamide is carefully considered in paediatric patients given the potential significant side effects of amenorrhoea and infertility. Ciclosporin and tacrolimus may be useful agents for refractory nephritis. Rituximab, an anti-CD20 monoclonal antibody, is typically used for refractory cytopenia and nephritis. Belimumab, a monoclonal antibody, requires individual TGA approval and is only used in a tertiary subspecialty setting. Patients receiving disease-modifying agents for cSLE require regular blood monitoring to avoid toxicities.
Other treatment strategies
Plasmapheresis and autologous bone marrow transplantation are options for severe, life-threatening disease.
Childhood-onset cSLE and COVID-19
COVID-positive paediatric patients with SLE who are taking immunosuppressive medications (high-dose corticosteroids, methotrexate, mycophenolate, azathioprine, B-cell-depleting monoclonal antibodies, calcineurin inhibitors) at moderate-to-high doses may be eligible for additional therapies including remdesivir, while the combination preparation nirmatrelvir plus ritonavir can be used in patients 18 years of age and older. Early discussion with the rheumatology team is advised. Patients on B-cell-depleting monoclonal antibodies (e.g. rituximab) may be eligible for pre-exposure prophylaxis with the combination preparation tixagevimab plus cilgavimab.
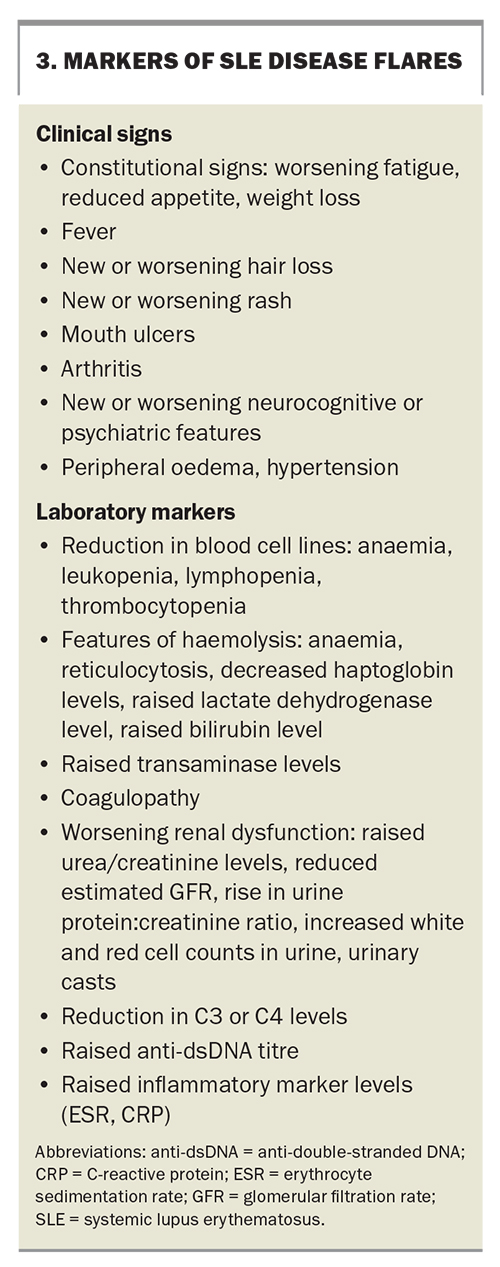
Recognition of disease flares
The early detection of disease flares is important. Features of disease flares are summarised in Box 3.
{kind=link}
Prognosis
Survival rates of patients with cSLE have significantly improved over the past several decades, with reported 10-year survival rates of up to 90% in developed countries.33,34 The causes of death in the paediatric population most often include infection, renal failure and cardiopulmonary disease. Patients may have significant morbidity from the prolonged use of high-dose corticosteroid therapy. Patients with cSLE have an increased risk of cardiovascular disease in adulthood.35
Conclusion
SLE with childhood onset is a complex disease process, requiring early diagnosis and initiation of treatment. The clinical presentation of SLE varies, making the diagnosis challenging for clinicians. Early communication between the general practitioner and rheumatology specialist is advised for all patients with suspected SLE. Management involves balancing the treatment of disease activity with medication toxicities. Further research is required to evaluate the efficacy and safety of novel therapies for cSLE. Education and support for the patient and family ultimately improves the outcomes and quality of life in patients with SLE. MT
COMPETING INTEREST: None.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.