Spinal muscular atrophy – a new treatment paradigm of proactive care
Spinal muscular atrophy (SMA) is a disease characterised by motor neuron loss. Disease-modifying therapies have transformed the approach to this progressive neurodegenerative disease into a new treatment paradigm of proactive care. Narrow therapeutic windows for intervention make timely diagnosis imperative to prevent irreversible loss of the motor neuron pool. Newborn screening for SMA offers a framework for achieving early diagnosis, improving equitable care and optimising health outcomes and cost-effectiveness.
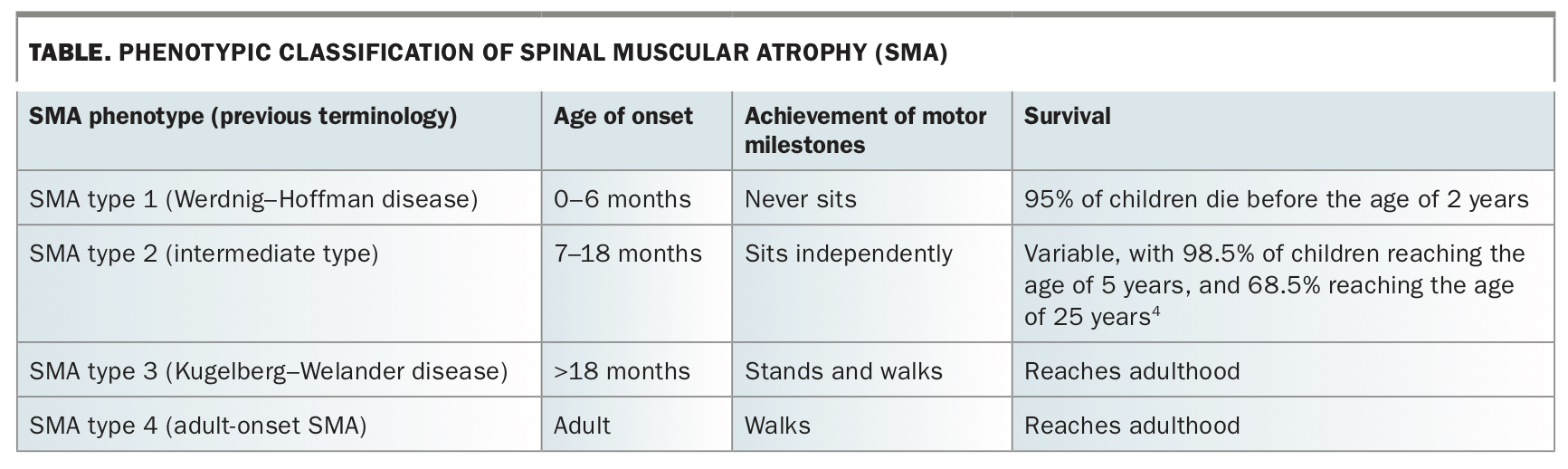
Spinal muscular atrophy (SMA) is one of the most common severe inherited disorders of infancy and early childhood, with an incidence of about one in 10,000 births and a carrier frequency of one in 45.1,2 SMA is characterised by muscle weakness from denervation of spinal and bulbar motor neurons.3 In its most common and severe form, SMA type 1, the natural history is characterised by respiratory failure and infant death. The wide phenotypic spectrum of the disease also includes SMA type 2, in children who manifest symptoms after the age of 6 months and can sit but never walk, and SMA type 3, in those who achieve independent walking (Table).5,6 People with SMA experience significant ongoing comorbidities related to progressive weakness and loss of function, including compromised nutrition, musculoskeletal deformities (scoliosis and contractures) and respiratory impairment.7
{kind=link}
SMA is an autosomal recessive condition, where mutations in the survival of motor neuron 1 gene (SMN1) cause a deficiency in survival motor neuron (SMN) protein, which is ubiquitously expressed and required for the development and maintenance of a healthy motor neuron.8 A back-up gene, survival of motor neuron 2 (SMN2), produces about 10% of full-length functional SMN protein. Known to be the most important modulator of SMA phenotype, SMN2 copy number varies in humans and ameliorates phenotype in a dose-dependent manner.9 As it influences severity and timing of disease onset, there is prognostic value in determining SMN2 copy number in children being screened for SMA. However, genotype–phenotype correlation is not absolute. Epidemiological studies show that about 95% of people with two or fewer copies of SMN2 have SMA type 1, whereas 5% have SMA type 2 or 3. The presence of three SMN2 copies encompasses all major phenotypes of SMA.9
New therapeutic landscape with disease-modifying agents
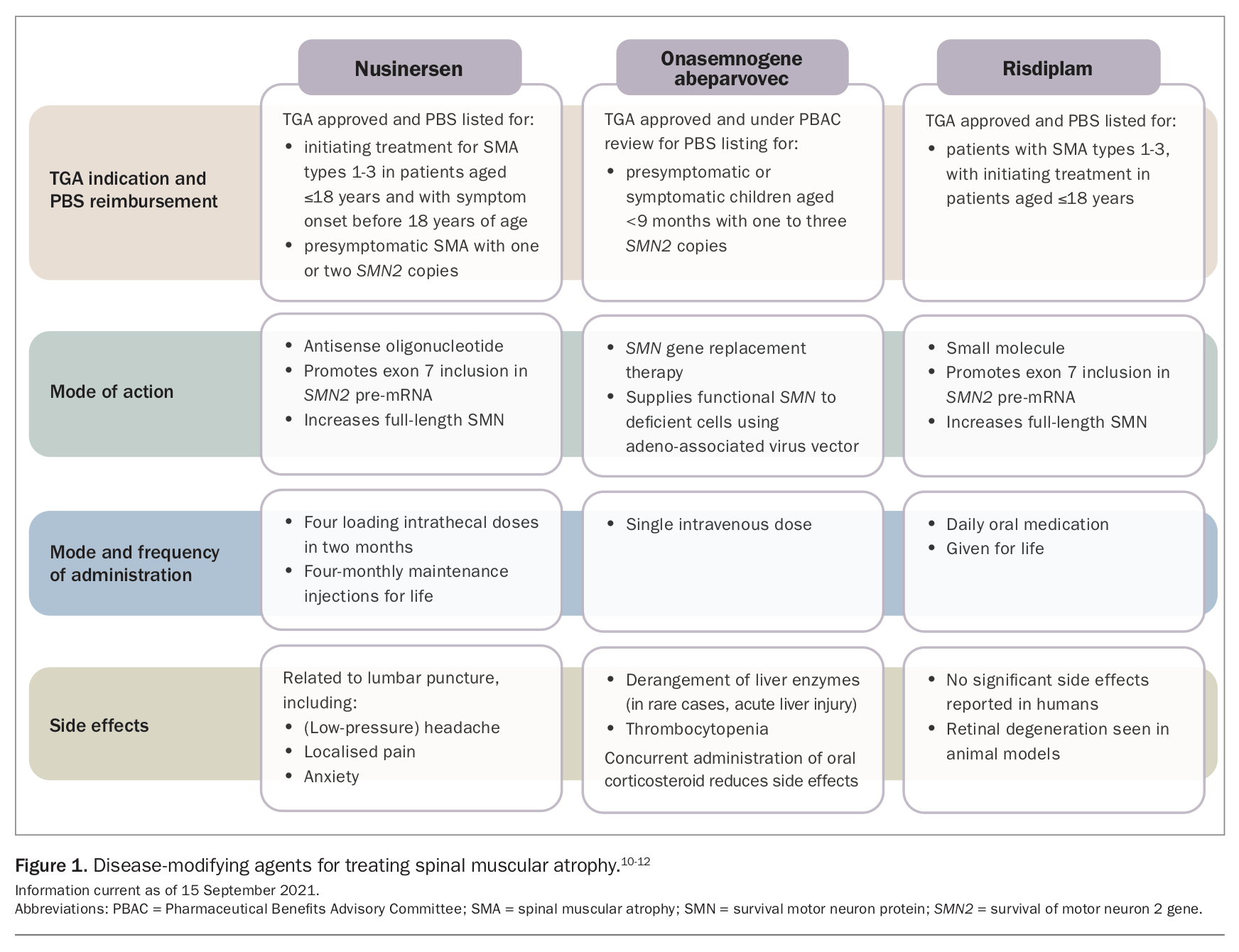
In 2018, nusinersen, an intrathecally administered antisense oligonucleotide, became the first disease-modifying agent for SMA to be approved by the TGA in Australia. Two further agents have since been added to the therapeutic repertoire, with each augmenting SMN protein levels (Figure 1).10-12 Onasemnogene abeparvovec, an intravenous adeno-associated virus gene therapy, was registered in 2021 for infants aged less than 9 months with symptomatic or presymptomatic SMA and with one to three copies of SMN2. The third therapeutic option is risdiplam, an oral daily small-molecule splicing modifier. PBS reimbursement criteria are related to age, genotype and phenotype and vary between each of these therapies.
{kind=link}
Clinical trials and early access programs for disease-modifying therapeutics have universally shown a significant improvement in survival for patients with SMA type 1.13 Early intervention is associated with clinically meaningful gains in motor function across all SMA phenotypes.13-17 Indeed, early access to intervention, and thus short disease duration, appears to be the major determinant of therapeutic response.18,19 To date, the most beneficial response to treatment has been seen in patients treated during the presymptomatic stage, with some infants achieving motor skills at developmentally appropriate ages.20 There is compelling evidence that, in patients with untreated SMA type 1, early and irreversible loss of motor neurons begins in the prenatal period, with rapid denervation in the first 3 months of life and 95% of the motor neuron pool lost by 6 months of age.21 However, the often subtle presentation of SMA type 1 in its early clinical evolution has led to a median delay of five months (range, 0.5–72 months) between symptom onset and diagnosis in Australia; a trend that has been replicated internationally.14 Longer delays of up to a year or more are seen in children presenting with milder forms of the disease.
Newborn screening: facilitating a shift to proactive care
To expedite initiation of treatment, the United States, Europe, Taiwan, Japan and Australia, among other countries or regions, have introduced newborn screening (NBS) programs or pilot studies for SMA.
The development of high-throughput genomic technologies and the ability to use them for screening purposes has made population-level screening for SMA feasible. Real-time polymerase chain reaction testing of the dried blood spot taken within the first few days of life has been used in several NBS programs to screen for homozygous exon 7 and/or 8 deletions in SMN1.22,23 In those with absence of SMN1 on the primary screening test, incorporating analysis of SMN2 copy number in a secondary screening tier helps clinicians delineate prognosis of the disease course and determines the avenues for treatment and monitoring.
As this assay does not detect 2 to 5% of SMA cases caused by other genetic mutation variants in SMN1 (including compound heterozygotes), it is important to maintain a clinical suspicion of SMA when a child presents with signs and symptoms consistent with disease onset. In clinical cases of SMA, recognition of the characteristic red flags continues to be essential (Box). In these cases, there should be a low threshold for expedient referral to specialist neuromuscular services, which can efficiently confirm the diagnosis and offer therapeutic intervention so the existing motor neuron pool can be salvaged.
{kind=link}
The Australian experience of newborn screening
In August 2018, a pilot NBS program for SMA began in New South Wales and the Australian Capital Territory to assess the feasibility, utility and public acceptability of NBS for the condition. The ongoing pilot was embedded into the existing NBS program and screened 202,388 infants in the first 24 months (personal communication, NSW NBS laboratory).
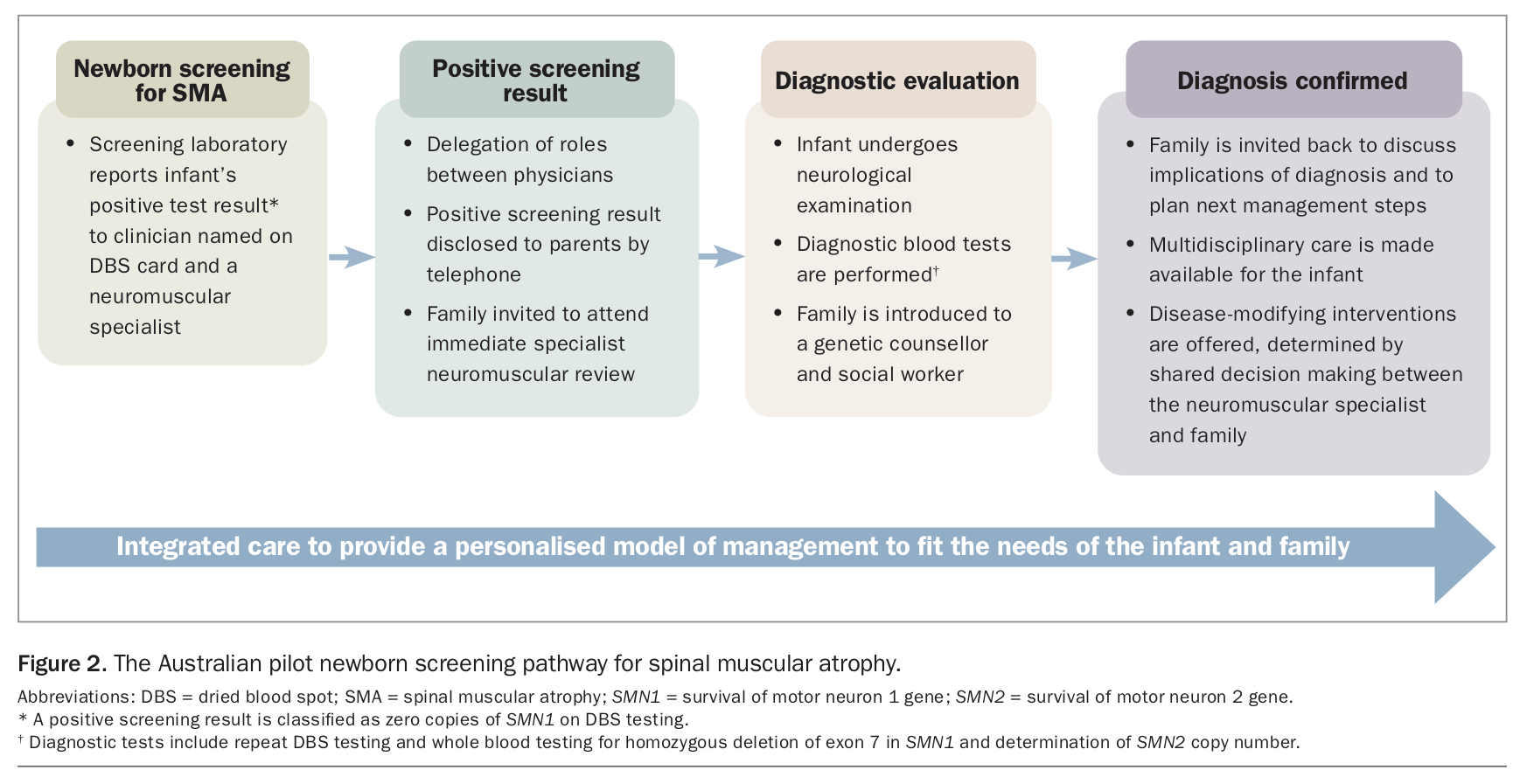
In the NBS program for SMA, the NBS laboratory reports a positive screening test result (i.e. an infant with SMN1 deletion positivity on the first dried blood spot) to the doctor designated on the dried blood spot card and a neuromuscular specialist (Figure 2). An assigned clinician then contacts the infant’s parents by telephone to disclose the positive result, and the family are invited to attend a specialist statewide neuromuscular centre for diagnostic blood tests and neurological assessment of the infant. Discussions about therapeutic planning, genetic counselling and multidisciplinary review follow diagnostic confirmation of the disease.
{kind=link}
The healthcare journey for many families with positive screening results in this program is strengthened by having access to local primary and secondary care physicians. Supported by neuromuscular specialists, clinicians in the community help resolve medical issues related to therapeutic intervention, facilitate discussions about the impact of the diagnosis on the family as a whole and guide appropriate access to local psychosocial support. GPs and paediatricians continue to provide shared care with specialist services, such as facilitating ongoing medical management of SMA-associated comorbidities, including monitoring of growth and development, maintaining immunisation schedules and implementing standards of care for affected individuals.
Data from the pilot have confirmed that NBS for SMA is sensitive and specific across the population.24 Encouragingly, results from the pilot indicate that the test is also acceptable to the population it serves.25 NBS for SMA promotes equity of access to diagnostic and care planning services and standardises the healthcare journey for affected children and their families.25 This is important for countering the geographical barriers, heterogeneous sociodemographic levels and variations in health literacy that are found in Australia. Routine national adoption of NBS for SMA would identify about 30 affected infants each year. Accordingly, in May 2021, the Australian Government Department of Health recommended that SMA be included in routine NBS programs in Australia, with individual state and territory governments managing implementation.
Expedient diagnosis as a gateway to care planning is expected to improve long-term health outcomes, not only by facilitating access to advanced therapies but as a pathway to supportive care, which remains the foundation of management. This includes access to measures such as respiratory support, nutritional supplementation, orthopaedic management and rehabilitation services, delivered within a multidisciplinary model of care and guided by recently updated standards of care, which are advocated for all patients.26,27 Findings from the pilot study have shown the clinical utility of this model of care, with a significant improvement in motor skills and a reduction in the need for ventilation and percutaneous gastrostomy feeding in those managed within the NBS for SMA framework, compared with historical symptomatic cohorts. Longer-term outcomes will be important in guiding future best practice.
Potential concerns about the inclusion of SMA in NBS include the high cost of ensuing treatment and the difficulties of sustaining access to therapeutics in health systems with finite resources. However, studies have found that NBS for SMA provides improved economic value for payers and patients when disease-modifying therapies are available.28
A further challenge of NBS for SMA is accurately predicting the timing of disease onset and severity of disease with the available prognostic biomarker of SMN2, as well as supporting families to make informed management decisions in light of this uncertainty. To resolve these evidence gaps, work is underway to identify biomarkers that can stratify patients based on disease severity and prognosticate disease progression, treatment response and intervention efficacy and durability.
Future of proactive care in Australia
A new treatment paradigm of precision medicine for SMA is unfolding and changing clinical practice to improve patient outcomes (see Practice Points). This model encompasses NBS for early identification and initiation of advanced therapies, but it also includes reproductive carrier screening for the condition. The aim of carrier screening is to advise couples of the risk of having a child with a specific disorder, allowing them to make informed reproductive choices and giving them access to prenatal and preimplantation diagnosis. This model acts as a primary prevention strategy to achieve population health gains through parents choosing to have a child without the condition or allowing timely postnatal treatment, thus targeting the narrow therapeutic window for the condition.29 In Australia, carrier testing can currently be accessed through commercial gene panels. However, studies are underway to ascertain the utility, feasibility and economic cost of pan-ethnic reproductive carrier screening for SMA and other life-limiting or disabling childhood-onset diseases, with a view to including such testing in the universal Medicare scheme.30
{kind=link}
Conclusion
Approved treatments for SMA have changed the natural history of the disease, and advances in NBS technology have made SMA an excellent candidate for inclusion in routine NBS programs. NBS for SMA may prevent the diagnostic odyssey met by many families, target a narrow therapeutic window and subsequently improve health outcomes for affected children. The Australian experience shows that population-level NBS for SMA is feasible, useful and equitable on a statewide level. Reproductive carrier screening may also be an important framework that underpins the new proactive paradigm of management for this condition. MT
COMPETING INTERESTS: Associate Professor Farrar has received speaker’s fees and personal fees for membership of the scientific advisory boards of Biogen, Roche and Novartis, outside the scope of the submitted work. Dr Kariyawasam has received speaker’s fees and travel support from Biogen, outside the scope of the submitted work.
ACKNOWLEDGEMENTS: Associate Professor Farrar acknowledges funding from the National Health and Medical Research Council of Australia: Investigator Grant (APP1194940). Dr Kariyawasam acknowledges funding from the Freedman Foundation and Research Training Program Scholarship, UNSW Sydney.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.