Newborn hearing loss: early diagnosis is vital

In Australia, three in every 1000 children are born with some degree of hearing loss. Early diagnosis is the key to early intervention and improved outcomes from hearing rehabilitation.
- Early diagnosis of hearing loss and intervention with appropriate hearing devices improves children’s language development as well as social, emotional and educational outcomes.
- All states and territories in Australia have universal newborn hearing screening (NBHS).
- Clinicians should have a high index of suspicion for hearing loss in children not meeting speech milestones or having behavioural problems and learning difficulties at school, and refer for audiological and/or ENT assessment.
- Children can have progressive hearing loss, so passing the NBHS does not mean a child will have normal hearing subsequently.
- GPs should discuss hygiene measures that can prevent the transmission of cytomegalovirus (CMV) infection with female patients who are pregnant and at higher risk.
- If an infant has a ‘refer’ result on NBHS, encourage screening for CMV via saliva polymerase chain reaction test within the first 3 weeks of life.
- CT scan is not an appropriate radiological investigation for newborn hearing loss; MRI of the brain and inner ear is preferred.
In Australia, one in every 1000 children is born with bilateral hearing loss in the moderate to profound range. Three children in every 1000 are born with some degree of hearing loss.
Most permanent hearing loss detected at birth (92%) is due to sensorineural causes including auditory neuropathy spectrum disorder (ANSD). A small proportion of cases (8%) are related to conductive hearing loss. Otitis media with effusion is a common cause of temporary conductive hearing loss in children and the most common cause of hearing loss worldwide. Congenital permanent conductive hearing loss can be due to abnormalities of the pinna, ear canal or middle ear resulting in microtia, canal atresia or ossicular chain abnormalities.
Early diagnosis of hearing loss and appropriate intervention are important in improving children’s language development and social and educational outcomes.
This article focuses on the investigation and management of sensorineural hearing loss.
Newborn hearing screening
Universal newborn hearing screening (NBHS) has revolutionised the diagnosis and management of newborn hearing loss. Testing is carried out using the automated auditory brainstem response (AABR), an electrophysiological test where electrodes are placed on the forehead and behind the ears of the sleeping baby. The results are binary: pass or refer (fail). A refer result requires a repeat AABR test within the next 48 hours. If the refer result is confirmed by the repeat test, then the newborn is referred for diagnostic audiology at a specialist centre (usually a children’s hospital).
Diagnostic audiology should be performed promptly after a refer result on the repeat AABR test and this can be carried out with the infant asleep, often after feeding. The diagnostic tests include acoustic auditory brainstem response, otoacoustic emissions and tympanometry.
Like any investigative tool, NBHS is not perfect. There are rare instances when hearing loss at birth may be missed or, more commonly, the child may have normal hearing at birth but progressive hearing loss over the following months and years.
Parents may notice children increasing the television volume or repeatedly missing verbal cues. Clinicians should have a high index of suspicion for hearing loss in children not meeting speech milestones, or having behavioural problems and learning difficulties in school or a diagnosis of attention deficit hyperactivity disorder. For these children, consider referral for audiological and/or ENT assessment as indicated.
Sensorineural hearing loss
The cause of hearing loss can be determined through a series of investigations including MRI scanning, blood and urine tests, as well as saliva tests for cytomegalovirus (CMV) by polymerase chain reaction (PCR). Supplementary investigations include ECG and ophthalmological examination when indicated.1
MRI of the brain, inner ear and internal auditory meatus is the most appropriate radiological investigation for newborn hearing loss. CT scan is not a first-line investigation and should not be ordered unless specified by an otolaryngologist or paediatrician with expertise in the assessment and management of newborn hearing loss.
With history, examination and investigation, an aetiology can be identified in two-thirds to three-quarters of children.
Causes of sensorineural hearing loss
Congenital bilateral sensorineural hearing loss is due to environmental, genetic and structural causes. Environmental causes account for approximately one-third of cases and include pre- and perinatal events such as congenital infections; extreme prematurity and risk factors associated with neonatal intensive care admission; ototoxic medications; meningitis; and jaundice requiring exchange transfusion. Congenital CMV infection is the most common environmental cause.
Genetic forms of hearing loss account for approximately half of bilateral sensorineural hearing loss. One-third of these will be syndromic (e.g. trisomy 21, Waardenburg syndrome, Usher syndrome). The remainder are nonsyndromic and mostly inherited recessively (85%). Up to 10% are transmitted in an autosomal dominant fashion and 2 to 5% through an X-linked or mitochondrial pattern of inheritance. More than 150 genes that cause hearing loss have been identified.
Structural causes of bilateral sensorineural hearing loss can be sporadic, environmental or genetic. Structural problems include cochlear dysplasia, cochlear nerve hypoplasia and large vestibular aqueduct syndrome.
In unilateral congenital sensorineural hearing loss, approximately 40% of children will have a structural abnormality on MRI such as cochlear nerve deficiency, cochlear dysplasia and enlarged vestibular aqueduct.2 Congenital CMV infection is another common cause. Genetic causes are uncommon in unilateral hearing loss and are usually syndromic (e.g. trisomy 21, branchio-oto-renal syndrome, Waardenburg syndrome).
Genetic causes of hearing loss
The most common genetic cause of hearing loss is connexin 26, which is most often inherited in an autosomal recessive pattern. This is usually an isolated genetic abnormality and, like most cases of genetic hearing loss, there is no associated syndrome.
Connexin 26 is a gap junction protein that plays an important role in potassium ion regulation in the organ of Corti in the cochlea. The gene encoding this protein is GJB2, located on chromosome 13. Geneticists have identified nearly 100 different mutations of GJB2, the most common being c.35delG, which is carried by 2 to 4% of the Caucasian population.3 The prevalence and specific mutations vary between ethnic groups.
Connexin 26-related hearing loss can be mild to profound. At least 25% and up to 50% of cases are progressive with deteriorating thresholds over time.3 Diagnosis is made via genetic testing on blood, which is a routine part of the diagnostic work-up for newborn bilateral sensorineural hearing loss.
Extended genetic testing is recommended as a second-tier investigation but due to cost it is not available in all centres. The most common gene panel for hearing loss is the OtoSCOPE, which tests for more than 100 genes associated with sensorineural hearing loss. When incorporating the OtoSCOPE panel into the routine diagnostic work-up for children with hearing loss, 75% will be given a definitive diagnosis of the cause of their hearing loss.
Infectious causes of hearing loss
Congenital CMV is the most common infectious cause of hearing loss and affects one in 150 newborns. At birth, CMV accounts for 10% of cases of hearing loss. This increases to 20% by 4 years of age.
Other infectious causes of newborn hearing loss include rubella virus, Toxoplasma gondii, Treponema pallidum and herpes simplex virus. These are extremely rare congenital infections in Australia because of high standards of health care and vaccination programs. Congenital rubella is more likely to present in a patient from overseas where there may not have been antenatal care or a vaccination program. In some Aboriginal and Torres Strait Islander communities in Australia, higher rates of T. pallidum infection increase the risk of congenital syphilis.
CMV is a herpes virus that is very commonly acquired in our community and is transmitted via saliva, urine or blood. By the age of 80 years, approximately 80% of people have evidence of prior exposure to CMV. When exposed, patients typically develop symptoms of a mild upper respiratory tract infection. CMV is commonly transmitted in a childcare environment and infected children can shed the virus in body fluids for months to years.
The Royal Australian and New Zealand College of Obstetricians and Gynaecologists recently released guidelines that recommend hygiene measures to all pregnant women in order to prevent CMV infection (Box).4 Previous CMV infection is not necessarily protective because different strains of CMV exist and CMV can reactivate during pregnancy. Most babies with congenital CMV are born to mothers experiencing a second infection or reactivation.
{kind=link}
In Australia, there is no universal newborn screening for congenital CMV. In some centres, infants who refer on newborn hearing tests have targeted screening for CMV via saliva or urine PCR tests. This testing needs to be completed by 3 weeks of age. After that time, it will be uncertain if the infection was congenital or acquired postnatally. Newborn screening blood spot CMV PCR can be helpful in the diagnosis but false negatives are an issue.
Early diagnosis can allow for treatment of moderate to severe congenital CMV infection soon after birth with oral valganciclovir over six months, which has been shown to reduce the severity of the disease.5
Auditory neuropathy spectrum disorder
ANSD encompasses a group of heterogeneous conditions that display similar characteristics on electrophysiological testing. Cochlear outer hair cell function is intact, reflected in present otoacoustic emissions or cochlear microphonic recordings. Auditory brainstem response testing is absent or markedly abnormal.
These results can be caused by abnormalities of the auditory pathway, anywhere from the inner hair cell to the cochlear nerve, brainstem and higher cortical pathways.
ANSD is found in 15% of newborns with permanent hearing loss, especially with prematurity, who may have experienced hypoxia, hyperbilirubinaemia and ototoxicity from antibiotics. Mutations of the OTOF gene, which encodes otoferlin, are most commonly associated with genetic ANSD.
Structural abnormality of the cochlear nerve including hypoplasia and aplasia can also result in ANSD. MRI is essential to diagnose these cases. Adults tend to have different causes of ANSD and are more likely to have an associated neurological condition or peripheral neuropathy.
Amplification of sound may benefit up to half of children with ANSD. Those failing to respond to early intervention amplification are offered cochlear implantation (Figure 1). Most patients with ANSD who undergo cochlear implantation have good long-term hearing and speech outcomes.
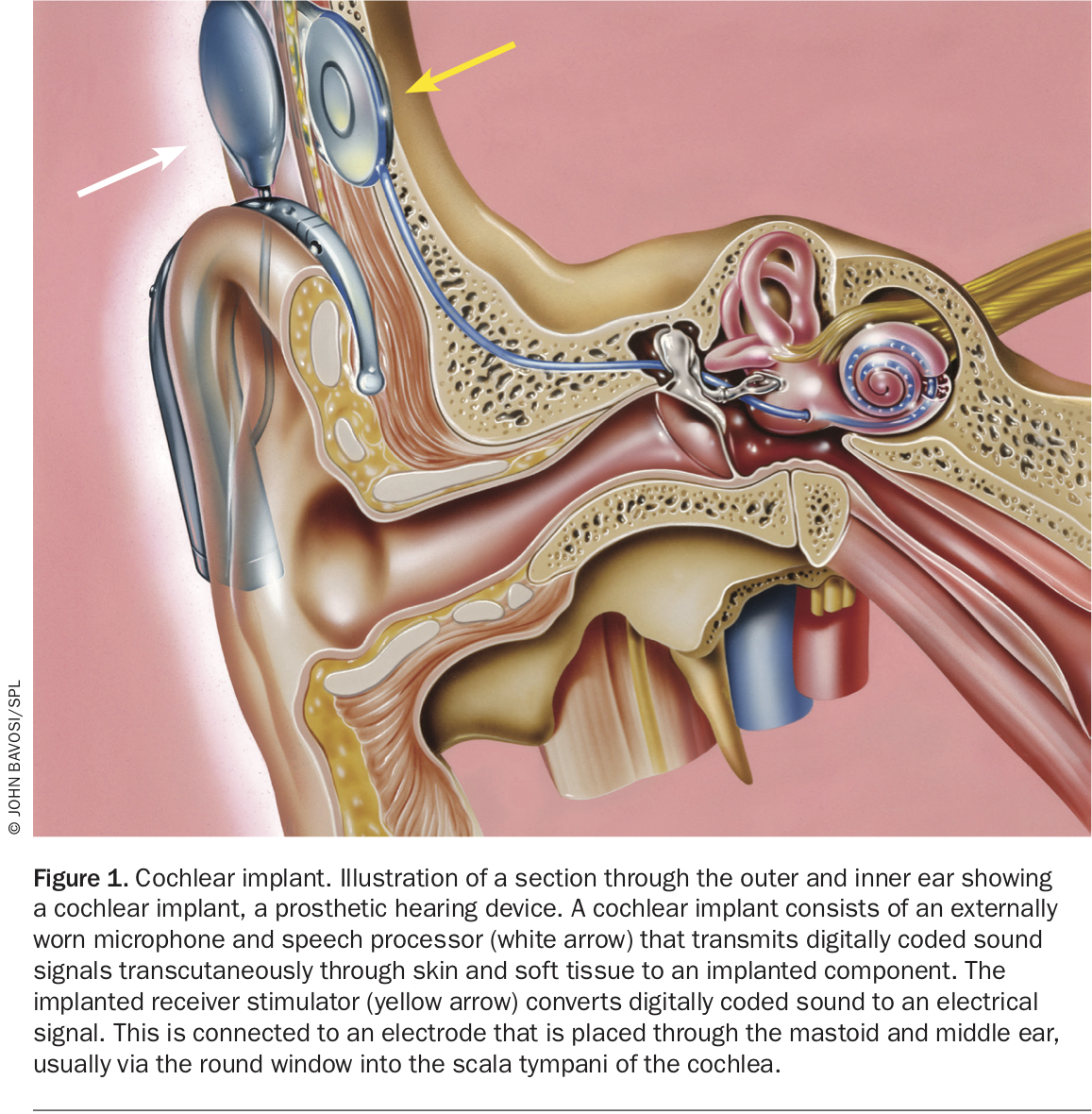{kind=link}
Large vestibular aqueduct syndrome
Large vestibular aqueduct syndrome (LVAS) is due to a structural abnormality of the temporal bone resulting in unilateral or bilateral widening of the vestibular aqueduct of the inner ear. The vestibular aqueduct is the bony channel for the endolymphatic duct, which is important for fluid and pressure homeostasis of the inner ear. Other anatomical abnormalities such as cochlear hypoplasia or enlargement of the semicircular canals or vestibule can occur in association with LVAS.
Patients with LVAS are usually diagnosed in the newborn period when they refer on NBHS and often have a moderate to profound hearing loss at the time of diagnosis (Figures 2a and b).
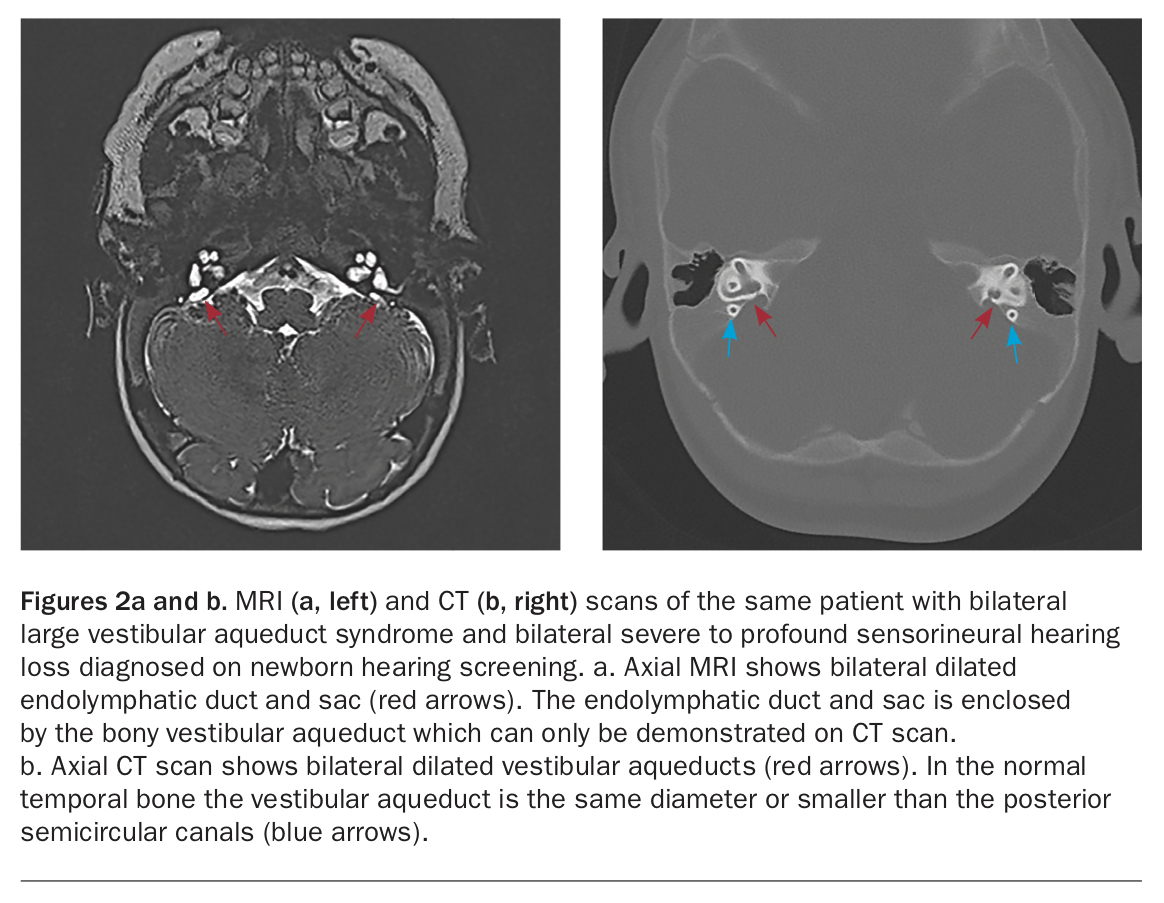{kind=link}
LVAS can occur as an isolated finding or associated with a genetic syndrome, most commonly Pendred syndrome; a triad of sensorineural hearing loss, goitre and defect in iodide organification which may result in hypothyroidism. Hypothyroidism can present at any age, but most commonly in the teenage years.
LVAS is characterised by stepwise deterioration of hearing associated with head trauma or barometric pressure fluctuations. Typically, a fall from one’s own height can result in hearing loss in patients with LVAS. Contact sport should be avoided. Prompt treatment with oral corticosteroids may prevent or defer hearing loss in the context of a sudden hearing decline.
Unilateral hearing loss
There has been a paradigm shift in the diagnosis, investigation and management of unilateral hearing loss over the past 10 years.
In the past, unilateral hearing loss was infrequently detected in childhood because most children developed speech normally. In cases of early diagnosis, parents were typically reassured that their child did not require investigation, intervention or follow up.
In the current era of universal NBHS, children are diagnosed with unilateral hearing loss at birth. There is increasing evidence in favour of early intervention in this cohort although many research questions still require answering. There is evidence that one-third of children with unilateral hearing loss have difficulty meeting educational milestones.6 More children with unilateral hearing loss are reported to repeat a grade at school and many have difficulty with sound localisation and understanding speech with background noise.
Approximately half of children with unilateral profound hearing loss will have an absent or hypoplastic cochlear nerve as the cause of their hearing loss. Children with unilateral profound hearing loss can be offered unilateral cochlear implants as there is research supporting earlier implantation (under 1 year of age) in this subgroup. Other options include a bone conduction device (on a headband or surgically implanted) or CROS hearing aid. For schoolchildren, an FM system/wireless communication device is helpful to hear the teacher over the background classroom noise.
Managing hearing impairment
Children who are diagnosed with hearing loss at birth are referred for early intervention. This involves regular audiology appointments for hearing aid fitting, and early intervention with teachers of the deaf and/or speech therapy.
Parents are offered referral to a paediatrician or ENT surgeon who can investigate the cause of hearing loss and help them access genetic testing and counselling.
Children with hearing loss in the severe to profound range, or those with ANSD who are not benefiting from hearing aids are referred for cochlear implant assessment.
Cochlear implantation as early as possible results in the best long-term speech and hearing outcomes for those meeting the criteria. For a child diagnosed with severe to profound sensorineural hearing loss at birth, the best outcomes occur when cochlear implantation is performed under the age of 12 months.
Children with underlying sensorineural hearing loss can develop acute otitis media and otitis media with effusion at the same rate as the rest of the paediatric population. It is important to manage middle ear disease effectively and consider early otolaryngology review in this cohort.
Longitudinal assessment of hearing is essential in all children with hearing loss, especially in cases of congenital CMV, LVAS and Connexin 26-associated disorders, all of which can be associated with progressive hearing loss. Hearing Australia (https://www.hearing.com.au) is federally funded to carry out regular audiological testing and, where indicated, hearing aid fitting and classroom modifications including wireless communication devices for children with hearing loss.
Conclusion
Universal NBHS means that most cases of hearing loss and impairment can be detected a birth. Sensorineural causes of hearing loss are most common and include environmental, genetic and structural causes. Early intervention including referral to an audiologist and/or ENT specialist is key to improved outcomes for hearing rehabilitation. MT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.