Systemic vasculitides. Part 2: Small vessel diseases
Small vessel vasculitides often manifest as neurological, renal, pulmonary and dermatological symptoms. Clinical features of systemic vasculitides are often nonspecific; therefore, it is important that GPs be aware of the range of symptoms that can help in the differential diagnosis, treatment and management of this diverse group of conditions.
- The two main categories of small vessel vasculitis are antineutrophil cytoplasmic antibody-associated and immune complex-associated disease, best diagnosed with renal and/or pulmonary biopsy.
- Severe renal failure and pulmonary haemorrhage are high-risk features that require urgent management.
- Eosinophilic granulomatosis with polyangiitis can be associated with severe cardiac disease including heart failure, conduction abnormalities and pericarditis. These complications account for half of the mortality associated with this disease.
- Hypocomplementaemic urticarial vasculitis and cryoglobulinaemic vasculitis are commonly associated with underlying systemic conditions including inflammatory rheumatological disease, chronic viral hepatitis and haematological malignancies. These should be screened for at the time of diagnosis.
- Leukocytoclastic vasculitis is the most common vasculitis encountered in clinical practice, only involves the skin and is most commonly associated with medication use.
- Patients receiving long-term corticosteroid or immuno-suppressive therapy require dedicated screening for chronic iatrogenic complications such as diabetes mellitus, osteoporosis, hypertension, obesity, opportunistic infections and secondary malignancies.
Part one of this two-part series on systemic vasculitides covered the large, medium and variable vessel vasculitides. We saw that most primary vasculitides are typified by nonspecific systemic manifestations such as fever, weight loss and lethargy but that classic presentations often aid our diagnostic efforts. We also stressed the importance of identifying high-mortality and high-morbidity situations such as visual impairment in giant cell arteritis (GCA) and coronary artery aneurysms in Kawasaki disease.
Part two will discuss small vessel diseases, a diverse group of diseases often with neurological, renal, pulmonary and dermatological manifestations, which are summarised in Table 1. We will conclude with a summary outlining a general approach to investigating and managing systemic vasculitides.
{kind=link}
Classification and clinical features
Antineutrophil cytoplasmic antibody-associated vasculitis
Granulomatosis with polyangiitis and microscopic polyangiitis
Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s disease, and microscopic polyangiitis (MPA) most commonly occur in patients over the age of 65 years and more often in Caucasian populations, affecting men and women equally. GPA affects the respiratory and glomerular epithelia producing haemoptysis and haematuria, respectively. Ear, nose and throat (ENT) manifestations such as sinusitis, otitis media, ulcers, polychondritis and subglottic stenosis are also classically seen. Dermatologic and ophthalmologic involvement can occur with variable frequency.
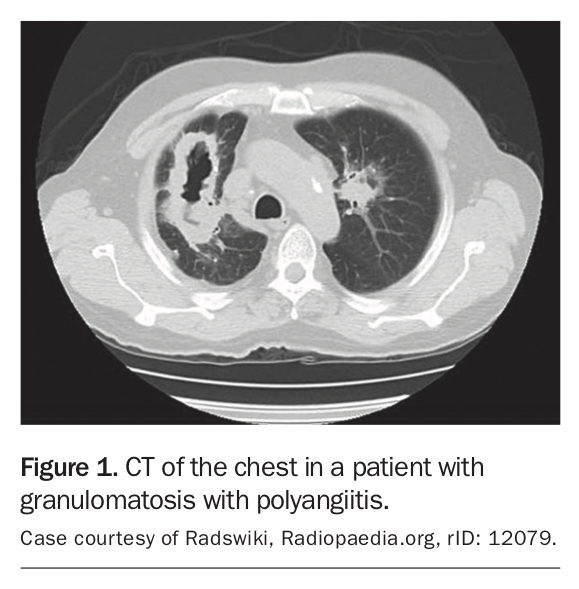
Cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) are seen in over 80% of patients with GPA and MPA and are typically associated with protease 3 in GPA and myeloperoxidase (MPO) in MPA.1 Urinalysis discloses haemoglobin and red-cell casts indicating glomerular inflammation. CT of the head, sinuses and chest is recommended in cases with ENT manifestations and pulmonary disease to clarify the site and severity of disease and guide biopsy (Figure 1).2 GPA and MPA are best diagnosed and differentiated on biopsy, usually renal or pulmonary, and defined by the presence of granulomatous inflammation.
{kind=link}
The goals of therapy are to reduce the incidence of renal failure and haemoptysis, and induce remission. Initial therapy begins with corticosteroids in combination with cyclophosphamide, rituximab or methotrexate for mild nonorgan- threatening disease. Plasma exchange therapy should be added in three situations:
- when patients have rapidly progressive renal impairment or renal failure at the point of requiring dialysis
- in those with pulmonary haemorrhage
- in the presence of concomitant anti-glomerular basement membrane antibodies.3
Maintenance therapy is started once corticosteroids are tapered, usually after three to six months, and preferred agents are azathioprine or methotrexate. Rituximab is an alternative agent for maintenance therapy but is not recommended for MPO-ANCA positive disease.4
Eosinophilic granulomatosis with polyangiitis
Eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as Churg-Strauss syndrome, is a multisystem vasculitis of the small and medium-sized vessel with gradual onset in the second or third decade of life. EGPA is typified by chronic rhinosinusitis, asthma and atopy in the context of peripheral eosinophilia. Atopic and eosinophilic phases of the illness generally predate nonspecific signs of systemic vasculitis such as fevers, weight loss and lassitude by as much as a decade. Cardiac involvement in the form of heart failure, conduction abnormalities or pericarditis accounts for roughly half of the mortality associated with EGPA. Mononeuritis multiplex is a classic neurological manifestation of EGPA and may be a clue to differentiate the disease from simple asthma or sinusitis in its early phase. Renal involvement is present in most patients with variable histopathology. Finally, it is worth noting that patients with EGPA are at a significantly increased risk of venous thromboembolism compared with the general population.5 Patients with EGPA have a raised peripheral eosinophil count, usually above 1500 mcmol/L, and roughly half have a positive peripheral antineutrophil cytoplasmic antibodies (p-ANCA). A case study on the diagnosis of EGPA is outlined in the Box.
{kind=link}
Chest imaging, especially CT, typically demonstrates transient patchy opacities, peribronchial and septal thickening, and helps guide bronchoalveolar lavage and pulmonary biopsy. Surgical lung biopsy revealing extravascular granulomas with eosinophilic pneumonia is the gold standard diagnostic test for EGPA and is superior in demonstrating typical histopathology to a transbronchial approach.6 Spirometry and pulmonary function testing typically demonstrate obstruction, with asthmatic-range reversibility with decreased lung volumes and impairment of gas transfer. Electrocardiography, echocardiography and occasionally endomyocardial biopsy are prognostically useful tests for determining the extent of organ involvement and guiding therapy.7
Initially, treatment with corticosteroids achieves remission without the use of adjuvant therapy in the majority of patients with EGPA. Cyclophosphamide is added when severe multiorgan involvement is present. Maintenance therapy with azathioprine, methotrexate or leflunomide is reserved for severe disease without multiorgan involvement and is continued for 12 to 18 months.8 Anti-interleukin-5 monoclonal antibody therapy (mepolizumab and reslizumab) has shown promise in treating EGPA.9
Immune complex-associated vasculitis
Anti-glomerular basement membrane disease
Anti-glomerular basement membrane (GBM) disease, also known as Goodpasture’s disease, is a small vessel vasculitis characterised by the presence of anti-GBM antibodies, usually directed against the NC1 domain of type IV collagen found primarily in the renal and respiratory tracts.10 Acute renal failure is the most common presenting illness and roughly half of affected patients have clinical evidence of alveolar haemorrhage with cough, progressive dyspnoea or overt haemoptysis.11 Concurrent systemic vasculitis is suggested clinically by the presence of constitutional symptoms such as fevers, weight loss and malaise, and by the presence of positive ANCA, usually MPO-ANCA, which is found in up to a third of patients with anti-GBM vasculitis.12
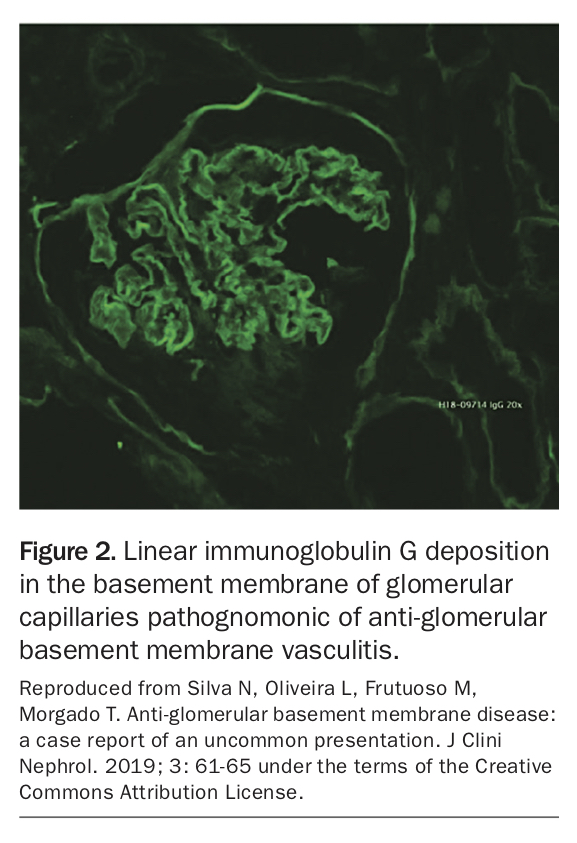
Demonstration of serum, pulmonary or renal anti-GBM antibodies using direct enzyme-linked immunoassay (ELISA) and confirmed with western blot is diagnostic. Renal biopsy should always be performed if possible as it is more accurate and guides treatment. Immunofluorescence of biopsy specimens reveals the pathognomonic linear immunoglobulin G (IgG) deposition in the basement membrane of glomerular capillaries (Figure 213).14
{kind=link}
Patients are managed initially with a combination of corticosteroids and either cyclophosphamide, rituximab or mycophenolate mofetil for three to six months, as well as up to six weeks of intensive plasmapheresis to remove circulating anti-GBM antibodies.15 Anti-GBM antibody levels, together with clinical response, guides the need for repeat plasmapheresis. Maintenance therapy with low-dose prednisolone with or without a second agent such as azathioprine or mycophenolate mofetil is recommended for nine months.
Immunoglobulin A vasculitis
Immunoglobulin A (IgA) vasculitis, also known as Henoch-Schönlein purpura (HSP), is a small-vessel vasculitis more common in Asian populations that predominantly affects children under 15 years old.16 The major clinical manifestations include palpable purpura primarily on the buttocks and lower limbs (Figure 3), non-deforming migratory oligoarthralgia of the lower limb large joints, colicky abdominal pain sometimes with bleeding and renal disease.
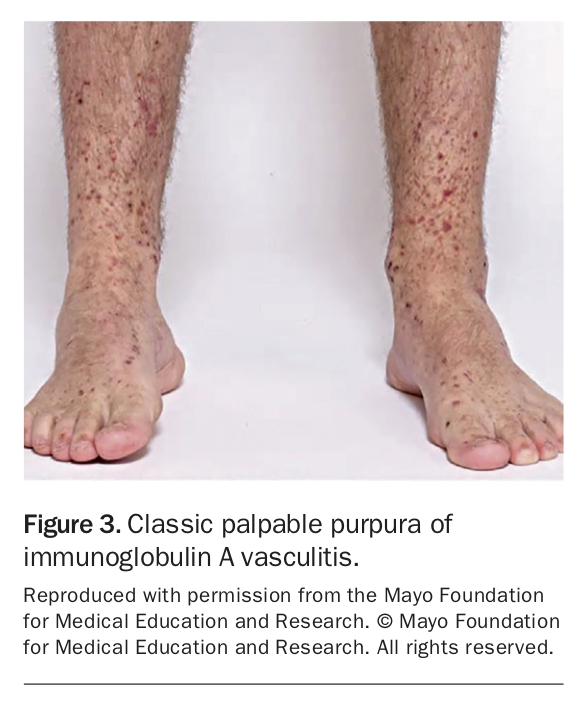{kind=link}
Haematuria and an active urinary sediment are typically found and renal dysfunction is typically self-limited.17 Markers of poor prognosis include prolonged abnormalities of the urinary sediment, heavy proteinuria, hypertension and impairment of renal function with an elevated serum creatinine level. Serum IgA levels are elevated in approximately half of cases and are associated with renal dysfunction.18 IgA deposition on immunofluorescence of skin or renal biopsy specimens is usually required to make the diagnosis in adult patients.
Hydration, rest and simple analgesia with NSAIDs are first line management strategies for symptomatic control. For patients with heavy proteinuria, therapy with an ACE inhibitor or angiotensin-receptor blocker (ARB) is recommended. Low-dose prednisolone may be considered in more severe or prolonged disease. Monitoring renal function and urinary sediment is a mainstay of management throughout.19 Patients with severe or prolonged disease (greater than six months) should undergo renal biopsy to demonstrate active inflammation and, if present, immunosuppressive therapy with cyclophosphamide or mycophenolate is recommended.20
Hypocomplementaemic urticarial vasculitis
Hypocomplementaemic urticarial vasculitis (HUV) describes a wide range of clinical syndromes characterised by the presence of urticaria and dermatological biopsy evidence of leukocytoclastic vasculitis. Immune complex deposition in vessel walls activates complement proteins, which stimulates mast cell degranulation and clinical urticaria.21 Complement protein levels are reduced in more severe or widespread disease. The clinical course is prolonged, usually occurring over months to years. Roughly half of patients with HUV experience constitutional symptoms and the most common sites of extracutaneous manifestations are summarised in Table 2.
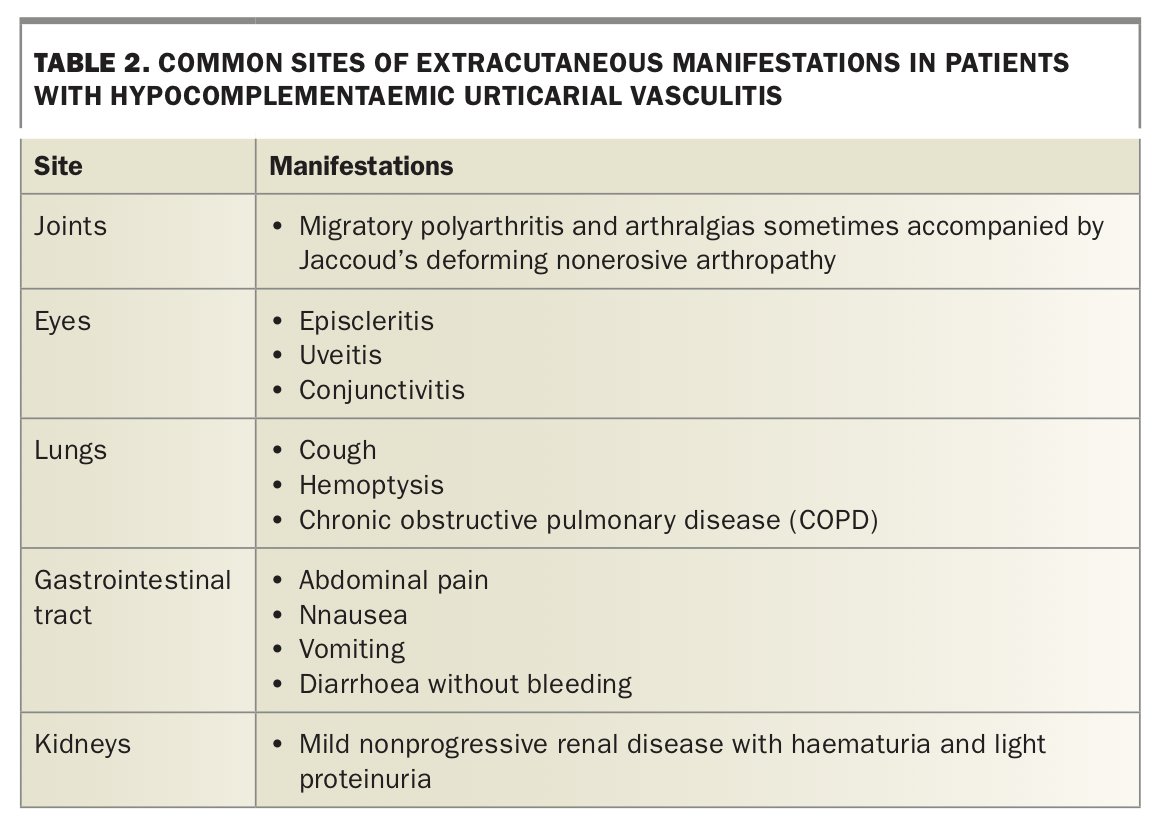{kind=link}
A skin biopsy should be undertaken early to guide further investigation and treatment. Should the diagnosis of HUV be confirmed on biopsy, inflammatory markers, renal function, urinalysis, hepatitis serology, ANCA and an autoantibody panel, complement protein levels with C1q precipitin (anti-C1q antibody), immunofixation electrophoresis and cryoglobulin levels should be obtained.
Most disease is mild and responds well to supportive treatment with antihistamines and NSAIDs. Corticosteroids with the addition of dapsone, colchicine or hydroxychloroquine may be indicated in cases where symptomatic treatment is inadequate. Severe disease, defined by the presence of end-organ dysfunction, typically requires longer term immunosuppressive therapy with mycophenolate mofetil or another steroid-sparing agent.24
Cryoglobulinaemic vasculitis
Cryoglobulinaemic vasculitis is a manifestation of cryoglobulinaemia, a heterogeneous group of diseases caused by the precipitation of blood proteins, chiefly immunoglobulin and complement. The systemic inflammation resulting from the precipitation of these proteins primarily affects medium and small vessels. Cutaneous manifestations are nearly universal.25 Arthralgias are also common with a proclivity for metacarpophalangeal and proximal phalangeal joints, knees and ankles.26 Peripheral neuropathy and renal involvement, primarily in the form of membranoproliferative glomerulonephritis, a subclinical small airways disease, also affects a sizable minority of patients with cryoglobulinaemia.
Investigations are aimed at diagnosis of underlying connective tissue disease (SLE, Sjögren’s), monoclonal haematological disorder (multiple myeloma, Waldenström’s macroglobulinaemia) or chronic viral infections (HCV or HBV). C4 complement levels are almost always reduced in cryoglobulinaemic vasculitis.27 Detection of cryoglobulins is a precise and complex laboratory process which must be executed correctly to avoid false positives. A cryocrit greater than 0.5% or cryoglobulin concentration greater than 50 mcg/mL is considered clinically significant.28 Management of cryoglobulinaemic vasculitis consists of identification and treatment of the underlying condition.
Leukocytoclastic vasculitis
Leukocytoclastic vasculitis (LCV), also referred to as hypersensitivity vasculitis, is the commonest vasculitis encountered in clinical practice. It is a skin-isolated vasculitis of the small vessels and may be idiopathic or secondary. The term ‘leukocytoclastic’ refers to the death and breakdown of neutrophils after degranulation. LCV presents most commonly with palpable purpura of the lower limbs without haematuria or other evidence of major visceral involvement. It is important to exclude systemic vasculitis or infection in cases of suspected LCV. Diagnosis is made with a combination of clinical and skin biopsy findings and is described by the American College of Rheumatology criteria.29 These criteria are met if at least three out of five of the following are observed:
- age over 16 years old
- temporal relationship to new or changed medication
- palpable purpura
- maculopapular rash
- perivascular neutrophils and fibrinoid necrosis seen on skin biopsy.
Skin biopsy occasionally reveals eosinophils, which strongly suggest a pharmacological aetiology. The most common medications implicated are sulfonamides such as frusemide and thiazide diuretics, penicillin and cephalosporin antibiotics, allopurinol, and phenytoin.30 In most cases, management is supportive in addition to cessation of the offending agent if known and possible.
Investigations
Although clinical features of systemic vasculitis are usually nonspecific, symptoms such as haemoptysis, haematuria, renal failure, asthma and jaw claudication can help sharpen the differential. Likewise, features of systemic disease such as SLE, rheumatoid arthritis, HIV or infective endocarditis can be helpful in directing investigations and therapy. Initial laboratory tests should include inflammatory markers (erythrocyte sedimentation rate and C-reactive protein), as well as measures of organ function such as serum creatinine level and liver function tests. Serology for HBV, HCV, HIV and cryoglobulins is typically undertaken as well. Urine specimens should be sent for routine analysis, microscopy and examination of the sediment. Microbiological cultures, serology and imaging guided by symptoms should be obtained to exclude simple infections. A careful medication and exposure history should be undertaken in consideration of a reactive process.
In the case of small vessel disease and/or vasculitis occurring in the context of another suspected systemic inflammatory condition, autoimmune serology should be obtained, including ANCA with protease 3 and MPO staining, antinuclear antibody, rheumatoid factor and complement protein levels. More specific serology should be performed to confirm diagnoses suggested by the initial screen, including anti-double stranded deoxyribonucleic acid antibodies, extractable nuclear antigens and anti-cyclic citrullinated peptide antibodies. In the presence of haemoptysis, plain chest radiography and high-resolution CT scan of the chest should be undertaken to guide subsequent therapy. In large vessel vasculitis and suspected polyarteritis nodosa, CT or magnetic resonance angiography is usually undertaken to visualise sites of involvement and confirm the presence of complications such as aneurysms, dissections or occlusions. Tissue sampling is often integral to diagnosis: temporal arteries in suspected GCA, renal biopsy in the context of renal dysfunction or an active urinary sediment, and skin biopsy with dermatological manifestations such as Henoch-Schönlein purpura or HIV.
Therapy and outcomes
Management of primary systemic vasculitides involves the induction and maintenance of remission, as well as prevention and management of complications and end-organ dysfunction resulting from vascular injury. Corticosteroids form the backbone of induction regimens, often in high doses compared with systemic rheumatologic conditions. In patients requiring prolonged maintenance therapy, steroid-sparing agents including thiopurines, mycophenolate and calcineurin inhibitors are preferred to long-term steroids. In secondary disease, management focuses on the underlying condition (i.e. cryoglobulinaemic vasculitis in HCV).
It is important to adhere to vaccination and infection prophylaxis guidelines, especially when combination immunosuppressive or high-dose corticosteroid therapy is used. Furthermore, patients with high burdens of corticosteroid therapy should be screened for systemic complications such as diabetes mellitus, hypertension, obesity and osteoporosis, and managed appropriately.
Diagnosis, management and monitoring take place primarily in the community, thus the general practitioner is integral to the management of most patients with vasculitis. Vasculitis accompanying systemic disorders such as rheumatoid arthritis or viral hepatitis often merit specialist involvement and regular follow up. Early and urgent referral to a vascular or other relevant physician specialist such as an immunologist or a rheumatologist should be obtained for patients who have evidence of progressive disease and in those with end-organ dysfunction. In patients with haemoptysis, severe renal impairment or suspected GCA with visual compromise, immediate hospitalisation is indicated.
Conclusion
The vasculitides are a diverse group of conditions whose specific manifestations generally depend on the size of vessels involved. Important complications of these varied diseases such as visual compromise, renal dysfunction, pulmonary haemorrhage and vascular aneurysms must be considered. Specialist involvement is crucial and should be obtained urgently if such complications are present. In patients receiving long-term corticosteroid or immunosuppressive therapy, dedicated screening for chronic iatrogenic complications such as diabetes mellitus, osteoporosis, hypertension, obesity, opportunistic infections and secondary malignancies is crucial to achieve optimal health outcomes. MT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.