A woman with diffuse skin-coloured nodules
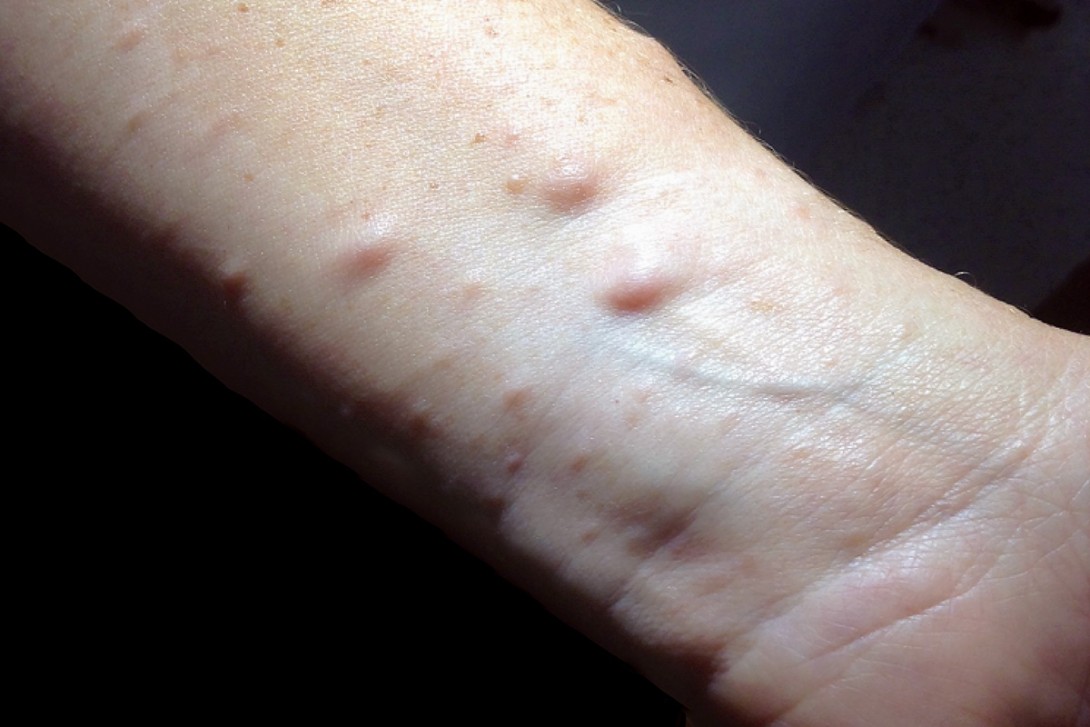
Test your diagnostic skills in our regular dermatology quiz. What is the cause of this patient’s skin-coloured nodules and other skin lesions and her occasional widespread pruritis?
Case presentation
History and examination
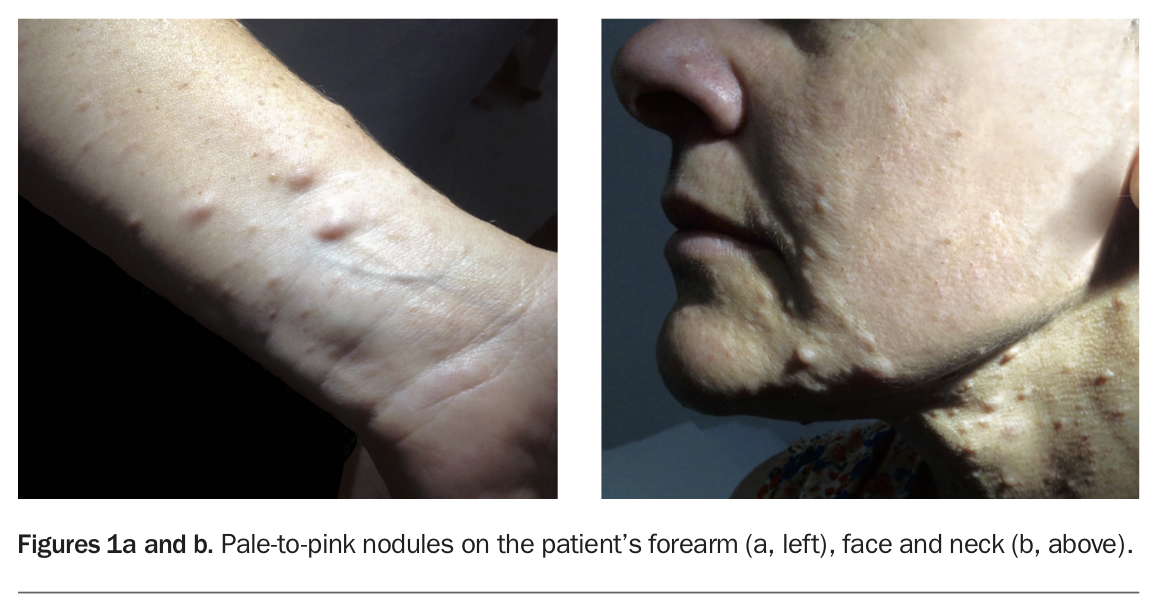
A 42-year-old woman presents with pale skin-coloured nodules on her left forearm that have developed over the last few years (Figure 1a). Similar lesions are present on her posterior trunk, face and neck, but she is unsure when these appeared (Figure 1b). She reports occasional pruritus, which is not always localised to the nodules and can be widespread. The lesions do not cause pain or dysesthesia and have not increased in size.
{kind=link}
The patient’s medical history includes hypertension, scoliosis, attention deficit hyperactivity disorder and learning difficulty. Her first period occurred before her eighth birthday. Her regular medications include perindopril and the oral contraceptive pill. She has no allergies. There is no family medical history of note.
On examination, pale-to-pink cutaneous nodules are noted on the volar surface of the forearm, the flexural surface of the neck and the perioral skin. In addition, there are multiple skin-coloured lentigines affecting the axillary folds and anterior chest, as well as large, uniformly pigmented, tan-to-brown patches (15 to 20 mm in diameter) with smooth borders involving the trunk and extremities.
Diagnosis and management
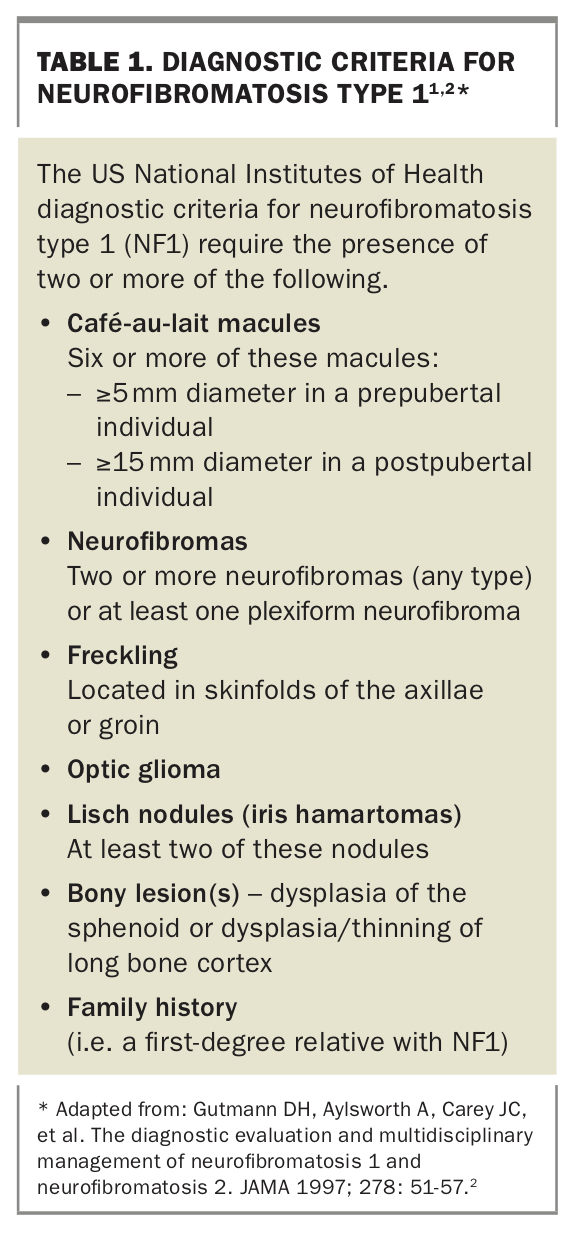
A skin biopsy was performed on the cutaneous nodules, which revealed the lesions to be neurofibromas. Ophthalmology review was arranged to assess for Lisch nodules (benign tumours on the iris), and a positive result returned. On the basis of these results, including the presence of more than two Lisch nodules, and the axillary freckling, the patient was diagnosed with neurofibromatosis type 1 (NF1), a multisystem disease. Criteria for diagnosis of NF1 are listed in Table 1.1,2
{kind=link}
The principles of management in this case are threefold: confirmation of the diagnosis and genetic counselling; management of widespread pruritus; and cosmetic management of the neurofibromas. She is referred to a clinic with expertise in neurofibromatosis for screening for extracutaneous manifestations of the disease, management of itch and laser treatment of neurofibromas.
Discussion
NF1 is one of the most common phakomatoses (neurocutaneous disorders of structures originating from the embryonic ectoderm), with a prevalence of about 1 in 3500 individuals.3 It is inherited in an autosomal dominant fashion: a mutation of the NF1 gene leads to loss of function of the tumour suppressor gene neurofibromin, leading to constitutive activation of the RAS-mitogen-activated protein kinase (MAPK) pathway.
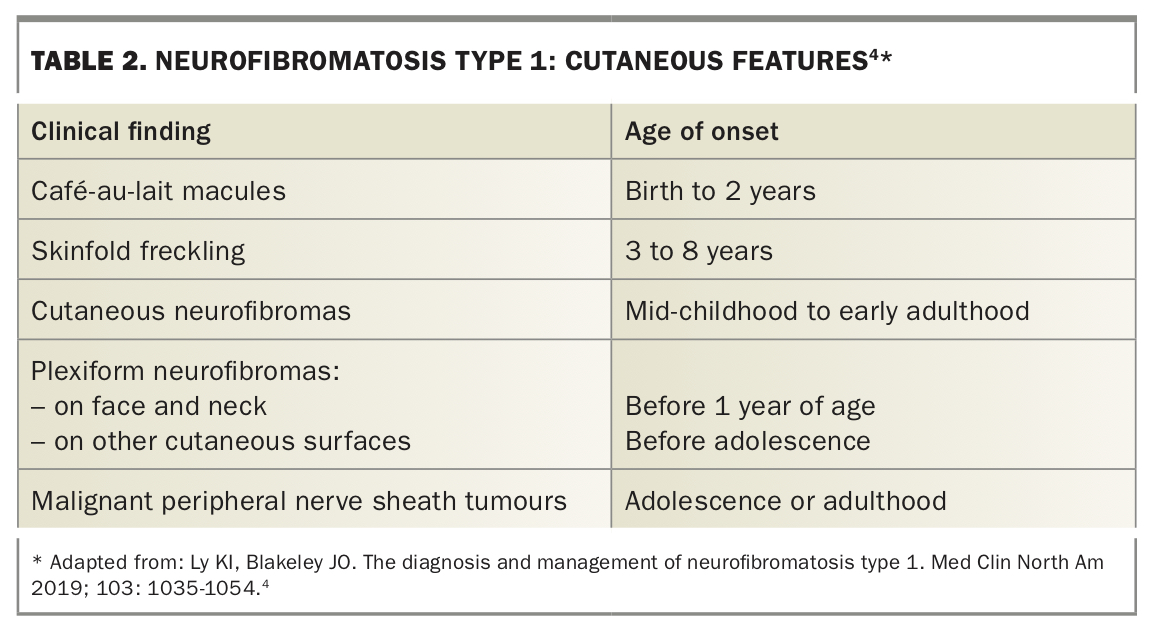
The age of onset varies for cutaneous features of NF1 (Table 2).4 Café-au-lait macules (CALMs) are one of the characteristic findings in NF1, with about 99% of patients exhibiting these as infants. CALMs characteristically appear as hyperpigmented brown patches with a smooth border. Axillary skin freckling is highly specific for NF1 – small skin-coloured freckles, up to 3 mm in diameter, begin to appear at around 3 to 5 years of age and involve the axillae, anterior flexural surface of the neck, inframammary skin and groin. The characteristic neurofibromas are composed of Schwann cells, mast cells, fibroblasts and collagen – they vary from soft, skin-coloured cutaneous neurofibromas to firm, nodular subcutaneous neurofibromas. Unlike CALMs and skin freckling, neurofibromas are not present in early childhood and tend to appear around puberty. Uncommon cutaneous findings associated with NF1 include juvenile xanthogranulomas, glomus tumours and naevus anaemicus and other vascular lesions.
{kind=link}
The extracutaneous manifestations of NF1 include optic glioma, gastrointestinal stromal tumours, neuroendocrine tumours (e.g. phaeochromocytoma), hypertension, scoliosis, astrocytoma, and malignant peripheral nerve sheath tumours. Itch is relatively common in patients with NF1 and thought to be neuropathic in origin.5
Differential diagnoses
Conditions to consider for a patient with a constellation of diffuse skin-coloured nodules, lentigines, uniformly hyperpigmented patches and bone deformity include the following.
- McCune-Albright syndrome (MAS). This rare multisystem condition is characterised by the triad of CALMs, fibrous dysplasia and endocrinopathies.6 It affects between 1/100,000 and 1/1,000,000 individuals. MAS is a mosaic condition caused by post-zygotic mutations in the GNAS1 gene, which confers a gain-of-function enabling constitutive activation of adenylate cyclase; this leads to excessive activation of G-protein-coupled receptors and downstream pathways and results in the over-production of multiple hormones.6 CALMs are the earliest sign, often present at birth or in the neonatal period, but can be missed because the lesions are frequently very light in colour. The CALMs in patients with MAS typically involve the posterior neck and lumbosacral spine and are less likely to involve the extremities – they are unilateral, have a sharp cutoff at the midline, and follow the lines of Blaschko. The irregular borders of CALMs in MAS are described as having a ‘coast of Maine’ jagged appearance; these are in stark contrast to CALMs in NF1, which have smooth ‘coast of California’ edges. MAS is a clinical diagnosis, usually made in the context of fibrous dysplasia (usually of the appendicular skeleton) and/or precocious puberty.6 Patients require a thorough assessment to screen for other systemic involvement of disease; investigations include imaging of the appendicular and axial skeleton for fibrous dysplasia, as well as screening for endocrinopathies (hyperthyroidism, hyperprolactinaemia, hyperparathyroidism, Cushing syndrome).
- LEOPARD syndrome. This genetic disorder, previously known as multiple lentigines syndrome, belongs to the group of neurocardiofacial-cutaneous syndromes.7 It is extremely rare – about 200 cases have been reported. ‘LEOPARD’ is an acronym for its most common clinical manifestations: (multiple) Lentigines, ECG conduction abnormalities, Ocular hypertelorism, Pulmonic stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness. It is inherited in an autosomal dominant fashion, with 85% of cases resulting from a missense mutation of the PTPN11 gene, which is implicated in the RAS-MAPK pathway.7 Brown-black lentigines appear at around 4 years of age, involving the face and trunk with sparing of the mucosa; there is a progressive increase in the number of lentigines from childhood (hundreds) to adolescence (thousands). The lentigines vary from small, discreet lesions measuring 1 to 2 mm in diameter to large CALMs. Unlike the early-onset CALMS in MAS and NF1, the CALMs in patients with Leopard syndrome are usually seen in adulthood. Screening for other components of the disorder, particularly the cardiac abnormalities, is required.7
- Legius syndrome. This rare, autosomal-dominant inherited disorder has an estimated prevalence of 1/75,000.8 It is caused by mutation of the SPRED1 gene, which results in dysregulation of the RAS-MAPK pathway.9 Patients with Legius syndrome develop CALMs (>90% of cases) and freckling (30 to 50%) but do not develop cutaneous tumours such as neurofibromas. However, neuropsychiatric disorders such as developmental delay, intellectual disability and behavioural disorders (e.g. ADHD) are shared between Legius syndrome and NF1.
Management
For a patient suspected of having NF1, the first step in management is referral to a genetic counselling service for confirmation of the diagnosis, consideration for molecular genetic testing and counselling regarding long-term management of the cutaneous and extracutaneous manifestations of NF1. Chronic pruritus, which can be widespread and often debilitating, is commonly a distressing problem for individuals with NF1 and is often managed with drugs usually used for neuropathic pain, such as doxepin.10 Neurofibromas require monitoring, particularly for areas of recalcitrant pruritus and/or pain, to exclude malignant peripheral nerve sheath tumour.3
Patients with NF1 require genetic counselling and lifelong monitoring. Many will live normal lives, but there are significant cosmetic issues for many and serious implications for some. MT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.