New hope for interstitial lung disease

Interstitial lung disease (ILD) comprises a large heterogenous group of disorders causing varying degrees of lung damage. Treatment options have historically been limited and are associated with significantly increased morbidity and mortality. This review summarises new advances in the diagnostic and management pathways of ILD.
- Interstitial lung diseases (ILDs) are a heterogenous group of conditions that cause lung parenchymal inflammation and fibrosis.
- Early recognition can be difficult but should be prioritised to allow rapid assessment with additional investigations.
- A multidisciplinary meeting discussion is key to establishing a diagnosis of ILD and, hence, an early referral to an ILD outpatient service or a local respiratory physician is critical.
- A comprehensive management strategy for patients with ILD requires a multidisciplinary team approach, utilising both pharmacological therapies and nonpharmacological measures.
Interstitial lung diseases (ILDs) are a heterogenous group of disorders that cause varying degrees of lung parenchymal inflammation and fibrosis. ILDs can be broadly categorised into two major groups. The first group are ILDs associated with an identifiable cause. These include occupational or environmental exposure (e.g. silicosis, hypersensitivity pneumonitis), drug-related ILD and ILD associated with an underlying connective tissue disease (CTD) such as systemic sclerosis (SSc), rheumatoid arthritis and dermatopolymyositis. The other major group encompasses ILDs with no clear predisposing factors or identifiable cause and are categorised as idiopathic interstitial pneumonia (IIP).1 Idiopathic pulmonary fibrosis (IPF) is the most common form of IIP, generally occurring in older adults, in which its incidence appears to be increasing. Other IIPs such as cryptogenic organising pneumonia, respiratory bronchiolitis-interstitial lung disease (RB-ILD) and rarer forms such as lymphoid interstitial pneumonia and idiopathic pleuroparenchymal fibroelastosis have also been described (Flowchart).
Establishing an accurate ILD diagnosis is important as ILDs have vastly different natural histories and prognoses. Furthermore, management of specific ILDs differs greatly, and with new therapeutic options becoming available, achieving an accurate diagnosis is paramount. However, identifying the exact aetiology can be challenging as symptoms are often vague and insidious in nature. Recently, new diagnostic approaches and pathways have been developed to improve the diagnostic accuracy of ILD. This article provides an overview of the diagnostic approaches and therapeutic advances in ILD – and demonstrates that there is, indeed, new hope for patients with ILD.
Diagnosis
Clinical history and examination
A thorough clinical history should be taken from any patient with a suspected ILD. A dry, unrelenting cough or exertional dyspnoea are commonly reported symptoms. A complete occupational and medication exposure history is essential as this may identify a cause for ILD and immediate action with drug cessation or antigen removal can be taken. A complete family history is also important to identify any history of pulmonary fibrosis.
Clinical examination often reveals fine crepitations when lung fields are auscultated. A complete rheumatological history and examination are essential to identify symptoms and signs of CTD such as Raynaud’s phenomenon, morning joint stiffness, arthralgias, rashes, myalgias or muscle weakness. Signs of right heart failure and cyanosis are also important to recognise.
What’s new?
Nailfold capillaroscopy (NFC) is a simple, noninvasive diagnostic tool that can be performed for patients with a suspected underlying CTD (Figure 1). Although described almost 100 years ago, its utility in detecting local microvascular changes that may support the presence of an underlying systemic CTD such as SSc is now recognised. As a proportion of patients with CTDs present initially with an ILD, the diagnostic role of NFC is now being explored in patients with undifferentiated ILDs. Furthermore, recent cohort studies have shown that capillary loss was associated with lower lung function, suggesting a prognostic role for NFC.2 Therefore, early referral to a rheumatologist to perform an NFC can be considered in patients with ILD, particularly if they have other symptoms suggestive of an underlying CTD.
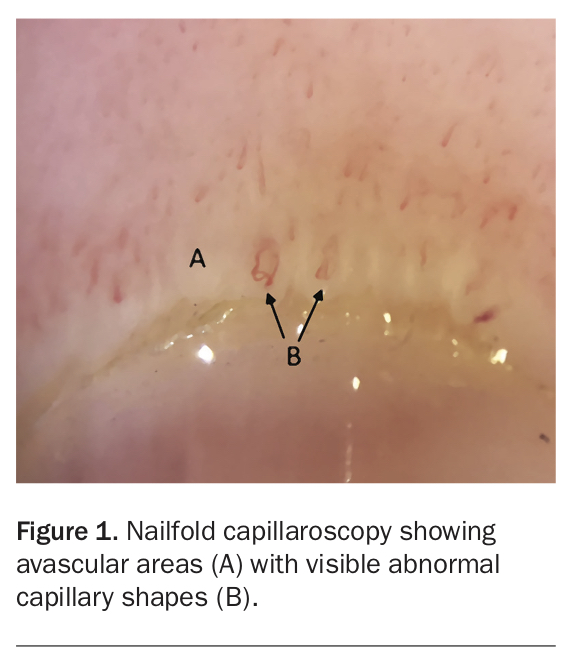{kind=link}
Pulmonary function testing
Bedside spirometry can often be performed as an initial assessment of the patients’ lung function. Reduced forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) with a normal or increased FEV1/FVC ratio is suggestive of a restrictive pattern, often seen in ILD. A complete pulmonary function test should be performed and will often confirm a reduced total lung capacity and a reduced diffusing capacity of the lungs for carbon monoxide. Pulmonary function testing is critical in diagnosing ILD and monitoring response to treatment.
What’s new?
The concept of a common progressive fibrosing phenotype of ILD, irrespective of the underlying diagnostic entity, is beginning to change the landscape of ILD management. Multiple cohort studies have shown that the health of a subgroup of patients with non-IPF ILD continued to decline due to progressive fibrosis despite treatment, with a natural history comparable with that of IPF.3-5 Patients with ILD should ideally have regular pulmonary function tests every three to six months. This not only helps to evaluate treatment efficacy, but also allows early recognition of disease progression and prompts further action if needed.
Autoimmune serology
Serological testing for autoimmune antibodies is recommended in the diagnostic workup of a patient with ILD. This assists the clinician in identifying underlying CTDs such as rheumatoid arthritis, Sjögren's syndrome, SSc and dermatopolymyositis, which are commonly associated with ILD. The exact list of serological tests needed has not been established and is dependent on available resources. A preliminary minimum panel should include an antinuclear antibody titre, rheumatoid factor peptide and anticyclic citrullinated peptide levels and an extractable nuclear antigen antibody panel. Measurements of C-reactive protein level, erythrocyte sedimentation rate and creatinine kinase level are also helpful to establish disease activity. Other additional autoantibodies like myositis-associated antibodies should be considered if a myositis-related ILD is suspected.
Radiology
A high-resolution CT (HRCT) provides critical radiological information needed for the diagnosis of ILDs and should be performed for every patient with suspected ILD. A recent international clinical practice guideline highlighted that, in the correct clinical context (i.e. in an older adult with no other identifiable aetiology), the presence of a usual interstitial pneumonia pattern on HRCT can be used to diagnose IPF (Figure 2).6 Meanwhile, the presence of a nonspecific interstitial pneumonia (NSIP) pattern in a younger patient with a known CTD is likely to carry a diagnosis of CTD-related ILD (CTD-ILD), which requires different management and has a different prognosis to other ILDs. Unfortunately, although various radiological patterns have now been described, there are a proportion of cases in which HRCT does not provide sufficient information for an accurate diagnosis of ILD (unclassifiable ILD).
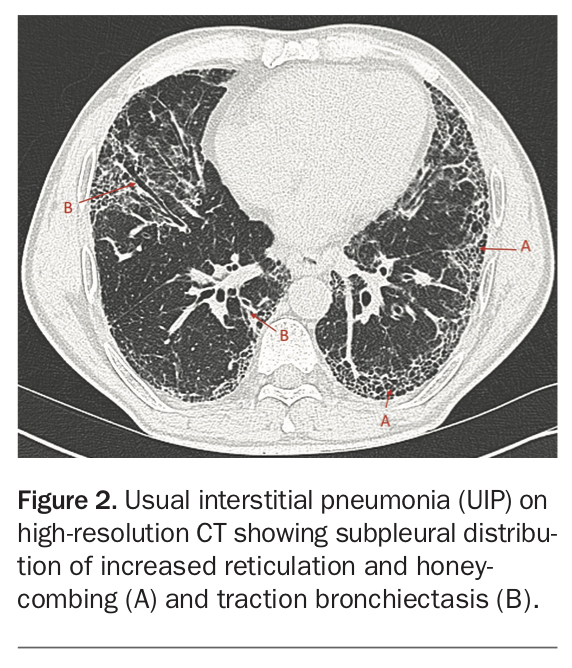{kind=link}
What’s new?
Quantitative CT analysis was designed to examine the density of discrete areas of the lung, providing a more accurate assessment of the severity of the underlying ILD. These analyses are potentially more precise and reduce interobserver variability among radiologists interpreting CT images. Studies have also shown a strong association between quantitative CT analysis data and FVC decline, compared with visual scores.7,8 These findings suggest that quantitative CT may play an important role in detecting disease progression. Although there are challenges in technique standardisation, it is predicted that quantitative CT will be incorporated into the management of patients with ILD by providing a complementary measure of disease severity and progression. With increasing interest in artificial intelligence in the form of deep and machine learning algorithms, future studies are likely to extend its use to quantitative CT in the development and validation of new diagnostic pathways to help distinguish ILD subtypes.
Histopathology
In cases where HRCT is not able to confirm an ILD diagnosis with high confidence, lung biopsy tissue may be required. Obtaining a lung biopsy, however, is an invasive procedure that is associated with significant complications such as bleeding, postoperative pain, exacerbation of the underlying ILD and death. Hence, any decision regarding lung biopsies should be made after a discussion with the patient and all relevant treating specialists.
What’s new?
Although the current standard practice of lung tissue sampling is performed through video-assisted thoracoscopic surgery (VATS), transbronchial lung cryobiopsy (TBLC) has emerged as a novel alternative, less invasive diagnostic technique. The COLDICE study was a recent Australian multicentre, prospective study that aimed to evaluate the accuracy of TBLC compared with VATS when used in patients with an unclassifiable ILD.9 The study showed that there was a high level of histopathological agreement and concordance between TBLC and VATS surgical lung biopsy. Although the study was not powered to assess safety, there were no new safety signals identified when compared with VATS. The study also highlighted the importance of a multidisciplinary approach, in both the decision-making process of performing such procedures and the interpretation of biopsy results.
Multidisciplinary meetings
The gold standard for ILD diagnosis is via a multidisciplinary meeting (MDM). The ILD MDM serves as a forum for clinical, radiological and histopathological data to be presented and integrated (Box 1). The main output of an ILD MDM is to provide a consensus ILD diagnosis and appropriate management recommendations. Key members who attend the ILD MDM include respiratory physicians, radiologists, histopathologists, ILD nurses, rheumatologists and immunologists.
{kind=link}
The utility of ILD MDMs have been demonstrated in multiple studies, where discussion at an ILD MDM resulted in higher diagnostic and prognostic confidence when compared with individual clinicians.10,11 However, the exact manner in which an ILD MDM is conducted has yet to be standardised and will likely depend on available local resources. Nonetheless, the Thoracic Society of Australia and New Zealand (TSANZ) has published a position statement advocating the pivotal role of ILD MDMs in the diagnosis of ILDs.12
In Australia, it is recommended that patients with a new ILD diagnosis be referred to the closest ILD outpatient service. The increasing use of telehealth services has been extremely useful for patients and physicians based in rural communities who do not have direct access to an ILD clinic. If direct access to an ILD clinic is unavailable, the patient should be referred to a local respiratory physician who can assist in referral to an ILD MDM.
What’s new?
It is now recognised that some patients present with clinical and serological manifestations of CTD but do not fulfil criteria for a defined rheumatological disorder. Idiopathic pneumonia with autoimmune features (IPAF) is the proposed standardised classification for these patients, although currently this terminology is only used as a research tool. However, recent studies suggest that a proportion of patients with IPAF will ultimately develop a defined CTD. Furthermore, patients with such CTDs may also first present with ILD as the only clinical manifestation of the condition without other extrapulmonary stigmata. Although myositis serology is not routinely used for screening, testing should be considered if there is a clinical suspicion for these conditions. An early referral to a rheumatologist or immunologist should be considered to help navigate the complexity of autoimmune and myositis serology testing.
Management
All patients with ILD should receive holistic care, using both pharmacological and nonpharmacological approaches. A multidisciplinary team of health professionals is usually needed for the management of ILD, with the general practitioner being the core member in the co-ordination of care. Although treatment options for ILDs have evolved significantly in the past few years, understanding and early discussions on the aims of treatment are critical in the management of patients with ILD. For example, the treatment goal for a patient with IPF is to slow the progression of disease, whereas the aim of treatment for a patient with CTD-ILD should be to stabilise the disease. Regardless of the approach, clear communication between health professionals is paramount for successful management.
Pharmacological agents
Antifibrotic medications
In 2014, two landmark randomised clinical trials were published on the antifibrotic agents, pirfenidone and nintedanib, for the treatment of IPF.13,14 Both agents were shown to reduce the rate of lung function decline and disease progression by around 50% in participants with IPF, a condition which previously had no available treatment. Post-hoc analysis of an open label extension study of pirfenidone showed the same treatment effects and safety profile in patients with more advanced disease. Similarly, the open label extension study of nintedanib showed a manageable safety profile over long-term use. Both pirfenidone and nintedanib are currently available through the PBS in Australia, but only patients with mild to moderate IPF can access these medications (Box 2).
{kind=link}
These agents have also been studied in other fibrosing ILDs. The SENSCIS trial investigated the efficacy of nintedanib in patients with SSc-associated ILD (SSc-ILD).15 This study showed that nintedanib slowed the annual rate of lung function decline compared with placebo, irrespective of background use of mycophenolate as an immunosuppressant. Another recent large multinational trial showed that nintedanib reduced the annual rate of FVC decline compared with placebo in patients with progressive fibrotic ILD of various aetiologies.16 A phase 2 clinical trial of pirfenidone in patients diagnosed with progressive and fibrotic unclassifiable ILD also suggests a benefit with an acceptable safety and tolerability profile.17 Currently, these agents are only approved for the treatment of IPF in Australia. However, these encouraging results provide much needed hope for these agents as potential future treatment for patients with progressive ILD despite optimal therapy.
Immunosuppression
Immunosuppressive agents are often used in patients with CTD-ILDs or cryptogenic organising pneumonia. Corticosteroids remain a mainstay agent, where initial high doses can be used in severe or rapidly progressive disease. However, monotherapy and prolonged use are generally avoided because of their significant side effects such as weight gain and osteoporosis. Other corticosteroid-sparing agents are often introduced to overcome these problems. Cyclophosphamide is a potent immunosuppressant that has demonstrated efficacy in improving lung function in patients with SSc-ILD but is limited by its potential toxicity. Mycophenolate mofetil is commonly used as maintenance therapy and has been shown to have similar efficacy and better tolerability compared with oral cyclophosphamide in SSc-ILD. Rituximab is a biological disease-modifying agent that is increasingly used to treat various CTDs and has shown some positive results in small cohort studies of patients with CTD-ILD. Although currently used as salvage therapy for patients who fail to respond to conventional treatment, there are clinical trials studying its role as a potential first-line agent.18
Immunosuppression is not indicated for all ILDs, with a previous study showing poorer outcomes when used in IPF. Furthermore, studies are often limited by the small participant numbers with no direct comparison data between agents. As there is no standardised management approach, early involvement of a physician experienced in ILD and a rheumatologist should be considered to help guide treatment decisions or any future changes to therapy.
Future novel agents
As our understanding of the pathophysiology of ILDs continues to increase, numerous potential therapeutic targets have been identified with novel agents at various stages of development. Recombinant pentraxin-2, a potent inhibitor of monocyte differentiation into profibrotic cells, showed positive results in patients with IPF when compared with placebo.19 A phase 2a trial of a novel autotaxine inhibitor (GLPG1690) showed promising results with a good safety profile and is now being studied in a larger trial.20 An inhibitor of galectin-3, a profibrotic protein, is also being studied in a phase 2b trial (ClinicalTrials.gov Identifier: NCT03832946). These are just a few of the increasing number of trials in ILD that continue to pave the way for a brighter future for patients with ILD (Figure 3).
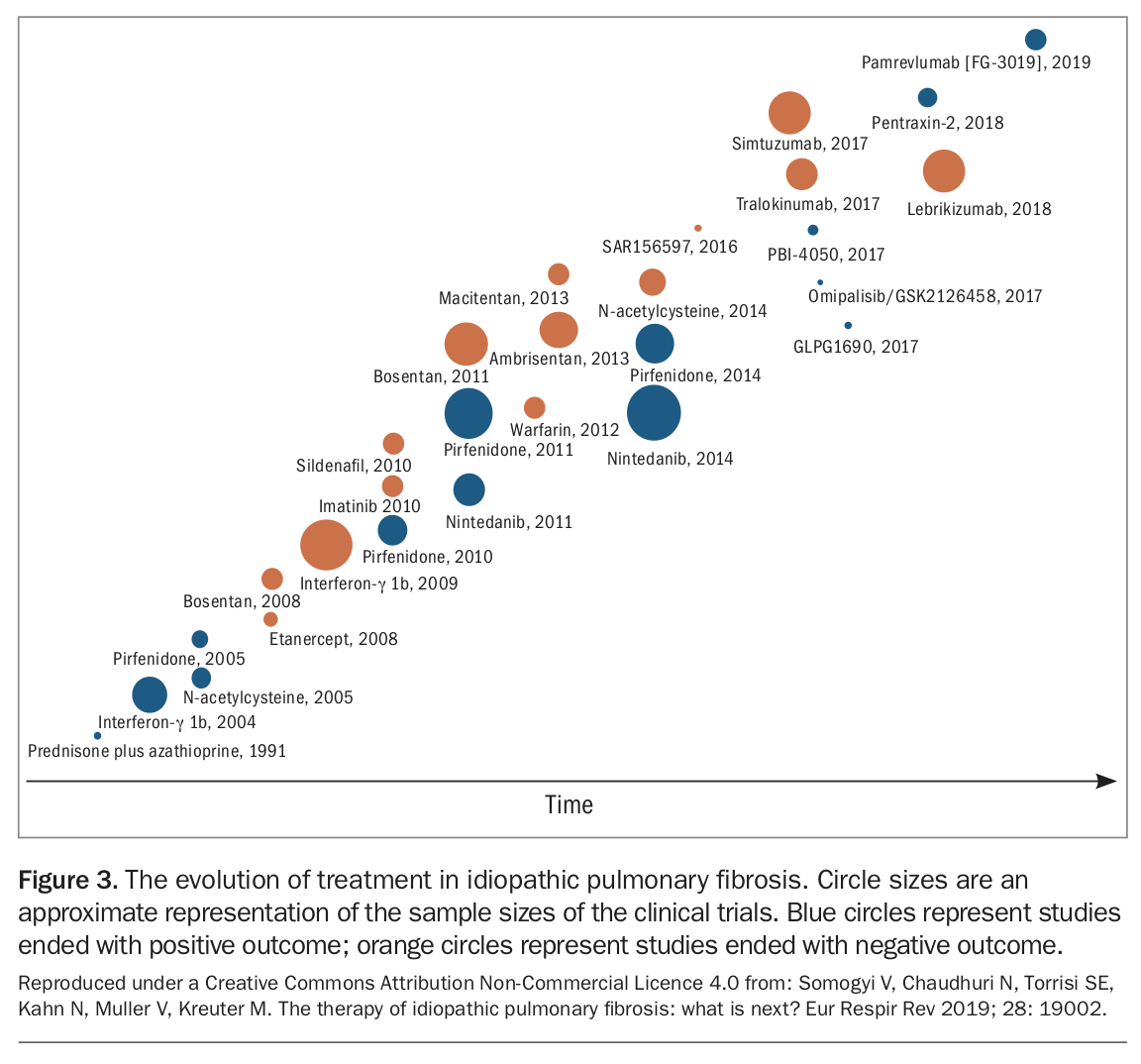{kind=link}
Nonpharmacological approaches
General considerations
Removal and avoidance of a known cause for ILD should be an initial approach to management. This is particularly important in drug-related ILD, hypersensitivity pneumonitis or ILD associated with occupational dust exposure. Smoking cessation is crucial and every smoker should be provided the necessary support, such as nicotine replacement therapy, to allow them to achieve that goal.
In light of the need for immunosuppression for some patients, it is important for patients to have their vaccination status updated to reduce the mortality and morbidity risk from vaccine-preventable diseases. As a minimum, patients should be offered the pneumococcal and annual influenza vaccines, both being inactivated (non-live) vaccines that are generally safe in patients receiving immunosuppressive therapy. These should ideally be administered at least two weeks before the commencement of immunosuppression. Live vaccines (such as the varicella vaccine) should be avoided if patients are already immunosuppressed and efforts should be made to administer them at least two weeks before treatment initiation.
Comorbidities
Comorbid conditions are common among patients with ILD and efforts should be taken to treat and manage them. Gastro-oesophageal reflux disease (GORD) is common in patients with IPF and SSc, often worsening their cough. However, current evidence remains unclear on the long-term safety of antacids in patients with ILD. Therefore, patients with ILD who also have GORD should be provided individualised treatment, combining lifestyle changes with antacids in select symptomatic cases.
Sleep disordered breathing is also a very common comorbidity, often causing nocturnal hypoxia, leading to secondary complications such as pulmonary hypertension and increased mortality. Patients with suspected symptoms of underlying sleep disordered breathing should be investigated with a sleep study and referred to a sleep physician.
Pulmonary rehabilitation
Pulmonary rehabilitation is available at most hospitals in Australia and usually entails an eight-week program of scheduled exercise sessions with physiotherapy guidance. These programs have shown significant benefits in improving symptoms and functional capacity in patients with ILD. There is also evidence of improvements in patients’ health-related quality of life. All patients with ILD should be offered an early referral to a pulmonary rehabilitation program at their local hospital.
What’s new?
Although there is increasing evidence that pulmonary rehabilitation can effectively address the symptom burden of patients with ILD, the optimal exercise regimen for ILD has not been fully explored. Preliminary evidence suggests that high intensity interval training (HIIT) is a well-tolerated training program that may be effective in improving exercise capacity in patients with ILD. Currently, an Australian multicentre randomised control trial is investigating the clinical benefits of HIIT compared with moderate intensity training programs (ClinicalTrials.gov Identifier: NCT03800914). Other recent advancements include rehabilitation programs conducted via teleconference. Although the effectiveness of telehealth in pulmonary rehabilitation is still an area of ongoing research, it allows patients in rural and regional settings access to these programs.
Oxygen therapy
Hypoxaemia in ILD is associated with significant morbidity and mortality. Supplemental oxygen therapy should be considered for patients with documented evidence of resting hypoxaemia (pO2 <55 mmHg or pO2 <60 mmHg in the presence of end-organ damage). These criteria may differ between states and local guidelines should be consulted. There is consensus among ILD experts that oxygen therapy should be considered in the treatment of nocturnal hypoxaemia in the absence of sleep disordered breathing. However, up to half of patients with ILD have exertional hypoxaemia in the absence of nocturnal hypoxaemia. Although current data suggest that ambulatory oxygen is beneficial in ILD, ongoing research is needed to provide more data on its clinical efficacy and a clinical trial is currently underway (ClinicalTrials.gov Identifier: NCT03737409).
Palliative care and patient support
An early referral to and involvement of a palliative care team is an essential part of a comprehensive approach to patients with ILD. A palliative care team’s main aim is to improve patients’ quality of life. Palliative care teams can use a variety of nonpharmacological strategies to alleviate symptoms of breathlessness. In certain cases, opioid medications can be used for patients with severe, refractory symptoms. However, care should be taken when using such agents and there should be clear communication and discussion among treating clinicians. In addition, a diagnosis of ILD is commonly associated with immense mental stress on both the patient and their family. It is important to offer a referral to a psychologist to provide an avenue for patients to address these issues. Numerous patient-support references are available for patients and their carers (https://lungfoundation.com.au/).
Lung transplantation
Lung transplantation provides a definitive cure for patients with ILD but is only suitable for a select few who fulfil strict selection criteria. In light of limited donor resources, it is important to consider the early involvement and referral of patients with ILD to a transplant centre for assessment by a lung transplantation team.
Conclusion
ILD encompasses a large number of different conditions that cause varying degrees of parenchymal damage, some of which leads to progressive lung function decline and eventual death. Early recognition and diagnosis of ILDs require input from a team of multidisciplinary health professionals, key among them being the patient’s general practitioner. Significant progress in new diagnostic methods, recognition of the role of early referrals to an ILD MDM and new therapeutic options provide invigorating hope for these patients. MT
COMPETING INTERESTS: None.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.