Genetic testing for epilepsy: why and when to think about it

Epilepsy is a common neurological condition associated with a number of comorbidities. By better understanding the underlying genetic causes of certain types of epilepsies, clinicians can provide patients and their families with clear information on the likelihood of the epilepsy being inherited, connect families with support groups and tailor health surveillance and therapeutic supports. Future advances in precision medicine may further improve health and psychosocial outcomes for people living with epilepsy.
Epilepsy is a common neurological condition affecting 65 million people worldwide, and thus is familiar to GPs.1 The impact of epilepsy on individuals and their families can be profound.2,3 Epilepsy is associated with a higher risk of premature mortality, with a twofold higher death rate among all patients with epilepsy in higher income countries; the death rate over 22-fold higher for patients with early onset seizures in the first year of life compared with the general population.4 Many epilepsies, especially those starting in early childhood, are associated with other health impacts, such as intellectual or learning disabilities, autism spectrum and sleep, breathing and gastrointestinal complications.5 Epilepsy is still a stigmatising condition in many cultures, and has profound impacts on mental health and wellbeing, not only for patients, but also for their carers and family members.3,6
Advances in genetic screening can help detect genetic causes of epilepsy. A genetic diagnosis is more likely when the epilepsy is severe, starting in the newborn or early childhood period, difficult to treat with first-line antiseizure medication, accompanied by other health or learning difficulties or affecting multiple family members. This article presents an overview of recent advances in our understanding of the genetic causes of epilepsy and provides a pragmatic, person-centred approach to management, including a clinical guide for when to consider genetic testing for epilepsy and which specialists to involve.
Acquired epilepsy
Worldwide, the leading diagnosable causes of epilepsy are acquired causes, such as hypoxia-ischaemia related to perinatal complications, cerebral infarcts or bleeds, tumours or cerebral infections including intrauterine ‘TORCH’ (toxoplasma, rubella, cytomegalovirus and herpes) infections and viral or bacterial meningitis and encephalitis.7 Assessment for acquired causes can be appropriately completed by taking a careful clinical history that includes antenatal and perinatal history and performing neuroimaging and infection screening.8,9
Genetic epilepsy
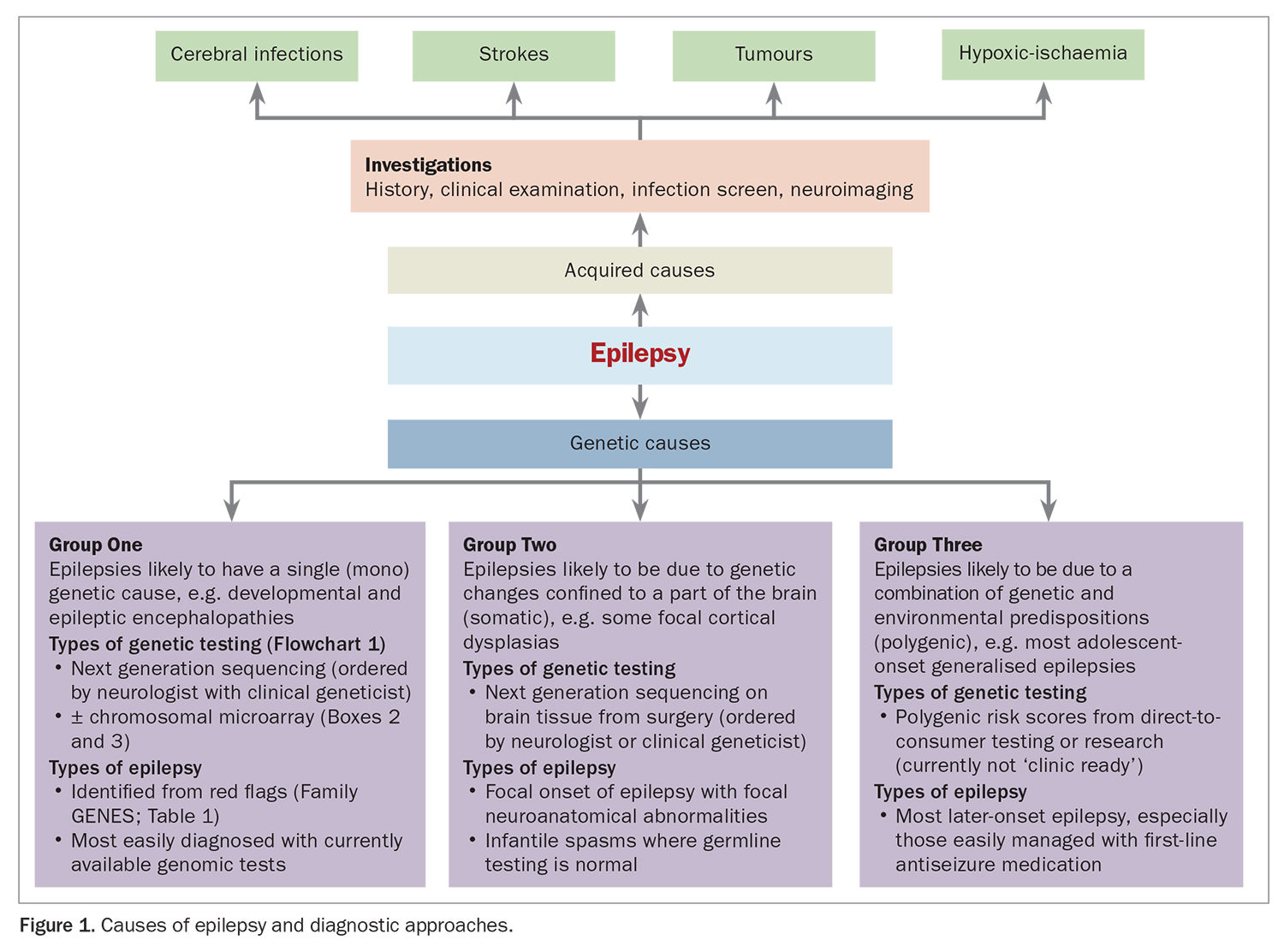
Recently, there has been a large shift in our understanding of the underlying causes of epilepsy.10 Genomic sequencing is being increasingly used to find genetic causes for some types of epilepsy, once acquired causes have been ruled out.3 Genomic testing is usually considered after an epilepsy diagnosis, based on the type and age of onset of seizures and accompanying comorbidities. When considering whether genomic testing may be useful, it is helpful to think of the range of genetic causes of epilepsy as falling into three distinct groups (Figure 1).
{kind=link}
Monogenic causes
The first group contains the monogenic causes, in which a ‘driving’ genetic change (an individual genetic or chromosomal pathogenic variant) can be identified. Over 600 genes and chromosomal regions have been linked to monogenic epilepsy. This is the group of genetic epilepsies that is most easily diagnosed with currently available diagnostic genomic tests.10,11 The mnemonic ‘Family GENES’ was developed to help primary practitioners consider which of their patients may have red flags for a rare genetic disease.12 This mnemonic fits epilepsy very well as shown in Table 1.
{kind=link}
The highest diagnostic yield is for the severe early onset developmental and epileptic encephalopathies (DEEs). DEEs are defined by the International League Against Epilepsy as the combination of severe epilepsy with an impact on development.13 Between 30 and 50% of children with DEEs have a monogenic germline cause found on current testing pathways.9
Somatic genetic causes
The second group contains somatic genetic causes, when a pathogenic genetic variant (or variants) is confined to certain cells, including in the brain, resulting in two distinct genetic cell populations.14 Termed ‘somatic mosaicism’, variants have been diagnosed by genetic testing of brain biopsies or surgically removed affected parts of their brain. Somatic variation is likely under-recognised in patients with severe intractable focal epilepsy. Research to improve the ability to diagnose somatic mosaicism in less invasive manners than surgery is ongoing.14,15 However, it is also important to recognise that some patients with severe focal onset epilepsy have a germline genetic cause; therefore, testing on blood or saliva should be done before considering genetic testing of brain tissue.16
Polygenic causes
The third group is polygenic causes, likely comprising a complex ‘soup’ of multiple genetic variants jostling together with environmental influences to tip an individual over into being more likely to develop epilepsy.17,18 It is likely that most people with adolescent- or adult-onset generalised epilepsies, such as juvenile absence and myoclonic epilepsies, have a polygenic cause for their epilepsy, if acquired causes have been excluded.19 Moreover, polygenetic influences may impact on exactly how monogenetic epilepsies present, explaining why two people with the same genetic variant may be differentially affected.17,20
Polygenic risk scores (PRS) are based on statistical analyses of genome-wide association studies and may suggest an individual’s genetic risk of developing a common condition.21 However, work is needed before these scores are ‘clinic ready’. Despite this, many direct-to-consumer companies offer PRS with a promise of determining the likelihood of a person developing a common condition and even their likely response to treatment. GPs may increasingly be presented with reports from these laboratories by their patients and should be aware that clinical and environmental factors are currently more accurate than PRS at estimating risk. Moreover, PRS may be falsely reassuring or report confusing false positives.21,22 Resources available to help guide GPs through potential minefields include a practical guide by Horton and colleagues,22 and the current NHMRC statement, which discourages the use of direct-to-consumer genetic testing (https://www.nhmrc.gov.au/about-us/resources/discussing-direct-consumer-genetic-dna-testing-patients). A clinical genetics service should be consulted if families are concerned about genetic risks and may be found through the NSW Health Centre for Genetics Education (https://www.genetics.edu.au/SitePages/Direct-to-consumer-testing.aspx).
Why think about a genetic diagnosis?
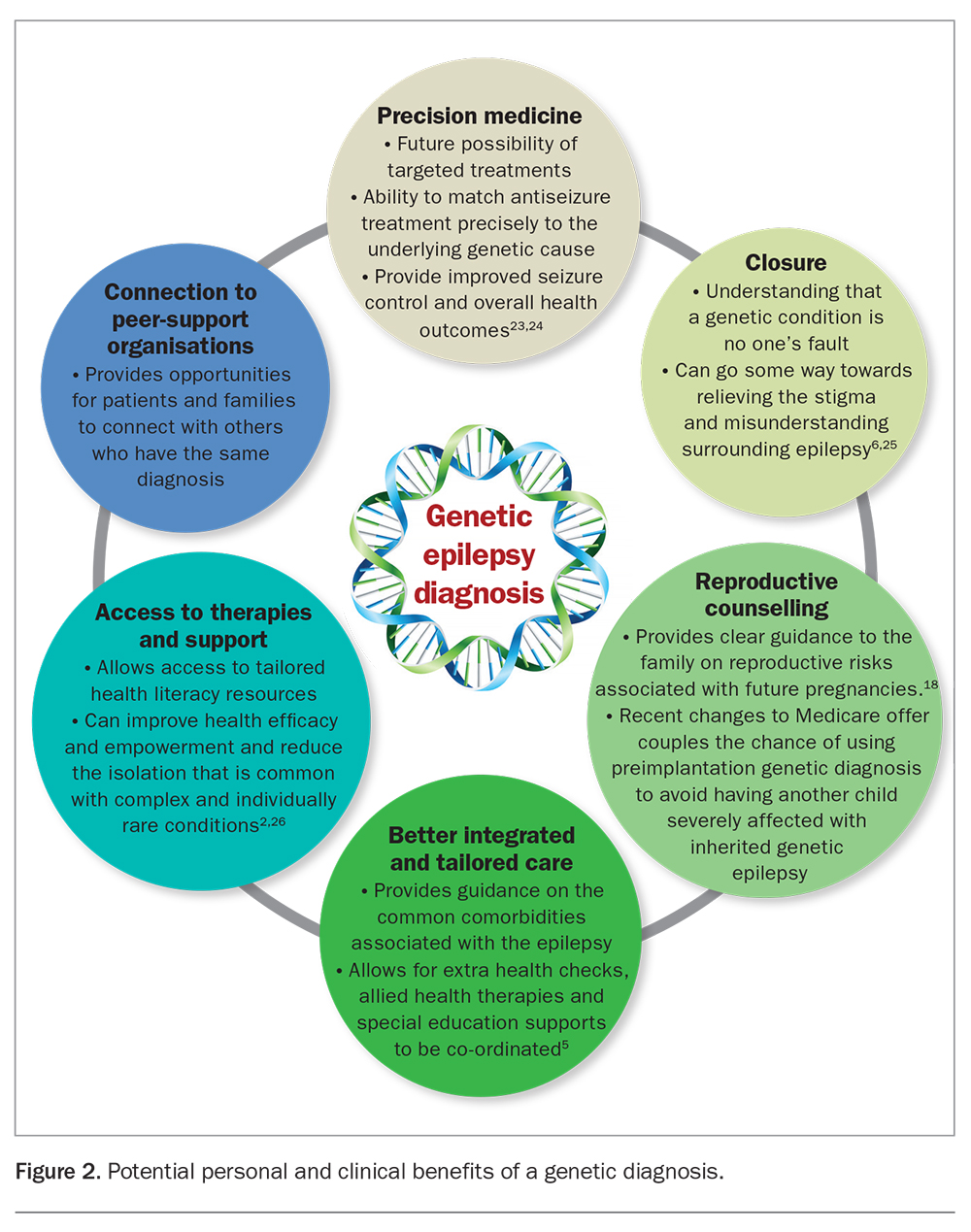
A clear-cut monogenetic or somatic genetic diagnosis can have many benefits for people with epilepsy and their families, as ummarised in Figure 2.2,5,6,23-26 However, genetic testing also has limitations and can create complications for patients and their families. Resources on genetic testing, including testing services, genetic counselling, informed consent, types of tests and shared decision making are listed in Box 1. Many genomic tests that are subject to a Medicare rebate require specialist paediatrician and clinical geneticists input for funding approval and for the laboratory to agree to conduct the test.
{kind=link}
{kind=link}
Informed consent process
A fully informed consent process outlining the potential benefits and complications is necessary before any genetic testing can be done.9,27,28 Clinical genetics services have experts in the informed consent process for genetic testing. Genetic Counsellors and Clinical Geneticists help guide families on shared decision making and provide pre- and post-test genetic counselling, which is needed to support the patient and family through the genetic testing process in a person-centred manner. The contact and referral details for clinical genetics services across Australia can be found on the Centre for Genetics Education website (https://www.genetics.edu.au/SitePages/Genetic-Services.aspx). GPs should also be aware of the informed consent process to help them discuss a referral to the genetics service with their patients and facilitate ongoing support for the patient and their family around the testing process and beyond.
Types of genetic testing
Some genetic tests can be organised directly by GPs. For example, a chromosomal microarray (CMA) can be organised under Medicare for certain indications (Box 2). As per the Royal Australian College of General Practitioners guidelines on Genomics in Clinical Practice, for individuals with epilepsy and developmental delay or intellectual disability, it may be helpful for a GP to order a CMA and a PCR test for Fragile X syndrome alongside a referral to a specialist. CMA is also an important first-line test for patients with possible ‘syndromic’ genetic epilepsy, such as for individuals with congenital anomalies or dysmorphic features in addition to their epilepsy (Flowchart 1). Such a pre-emptive approach can reduce the time to diagnosis for patients.27,29
{kind=link}
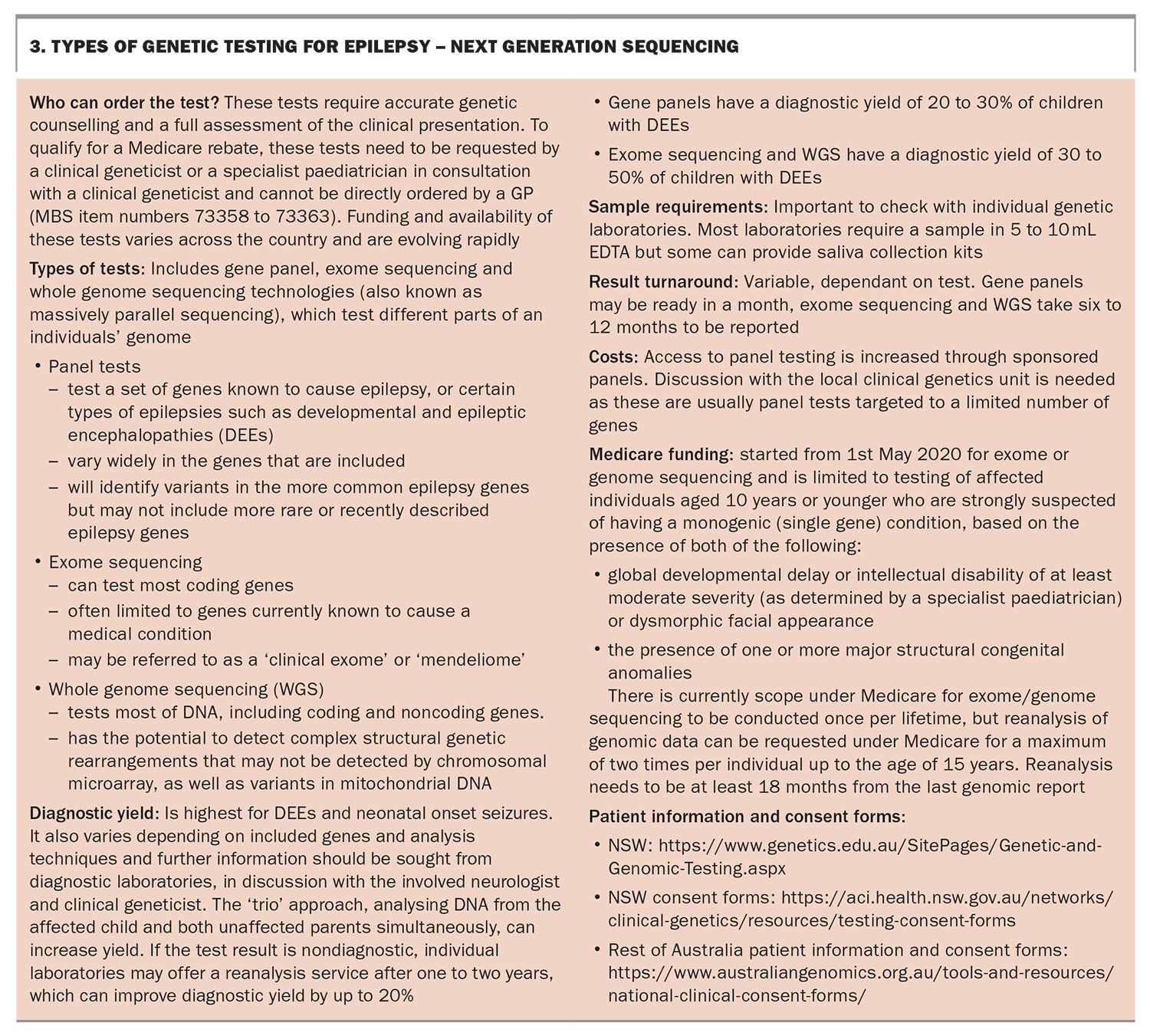
CMA is often completed before more detailed genomic testing with next-generation sequencing (NGS), which includes epilepsy gene panels, exome or whole genome sequencing. These genomic tests currently require specialist paediatrician and clinical geneticist input (Box 3). Although a full discussion of the advantages and practicalities of ordering each of these types of tests is outside the scope of this article, more information can be found from the resources in Box 1 and review papers.27,28,30 Key points on the types of genetic testing available for epilepsy are highlighted in Box 2 and Box 3.
{kind=link}
Shared decision making
The most important considerations about shared decision making in genetic testing for epilepsy are highlighted in Flowchart 2 and include the following.
- The potential for each test to find a clear diagnosis (expected diagnostic yield). This is best advised by the patient’s neurologist, potentially with input from a clinical geneticist, after careful profiling of the patient’s epilepsy. However, the expected diagnostic yield is rarely above 50% and often may be as low as 20 to 30%, depending on test and epilepsy type. The GP therefore plays an important role in setting accurate expectations from the start.
- The potential advantages of a diagnosis. The patient’s neurologist should be able to comment on the chance of a diagnosis altering patient management. It should be noted that there are not many truly targeted or curative therapies for genetic epilepsies, so expectations should be managed. However, this is certainly a hope for the future, and adiagnosis is a crucial first step to the later possibility of precision medicine.23,24 Currently recognised precision medicine for epilepsy include the use of sodium channel blockers, such as carbamazepine, for gain of function variants in SCN2A and SCN8A genes; avoiding sodium channel blockers for Dravet syndrome due to loss of function variants in the SCN1A gene; and use of the ketogenic diet for patients with GLUT1 deficiency due to loss of function variants in the SLC2A1 gene.23,24 The potential broader clinical and personal benefits of a diagnosis should be carefully explored with the patient and their family (Figure 2).
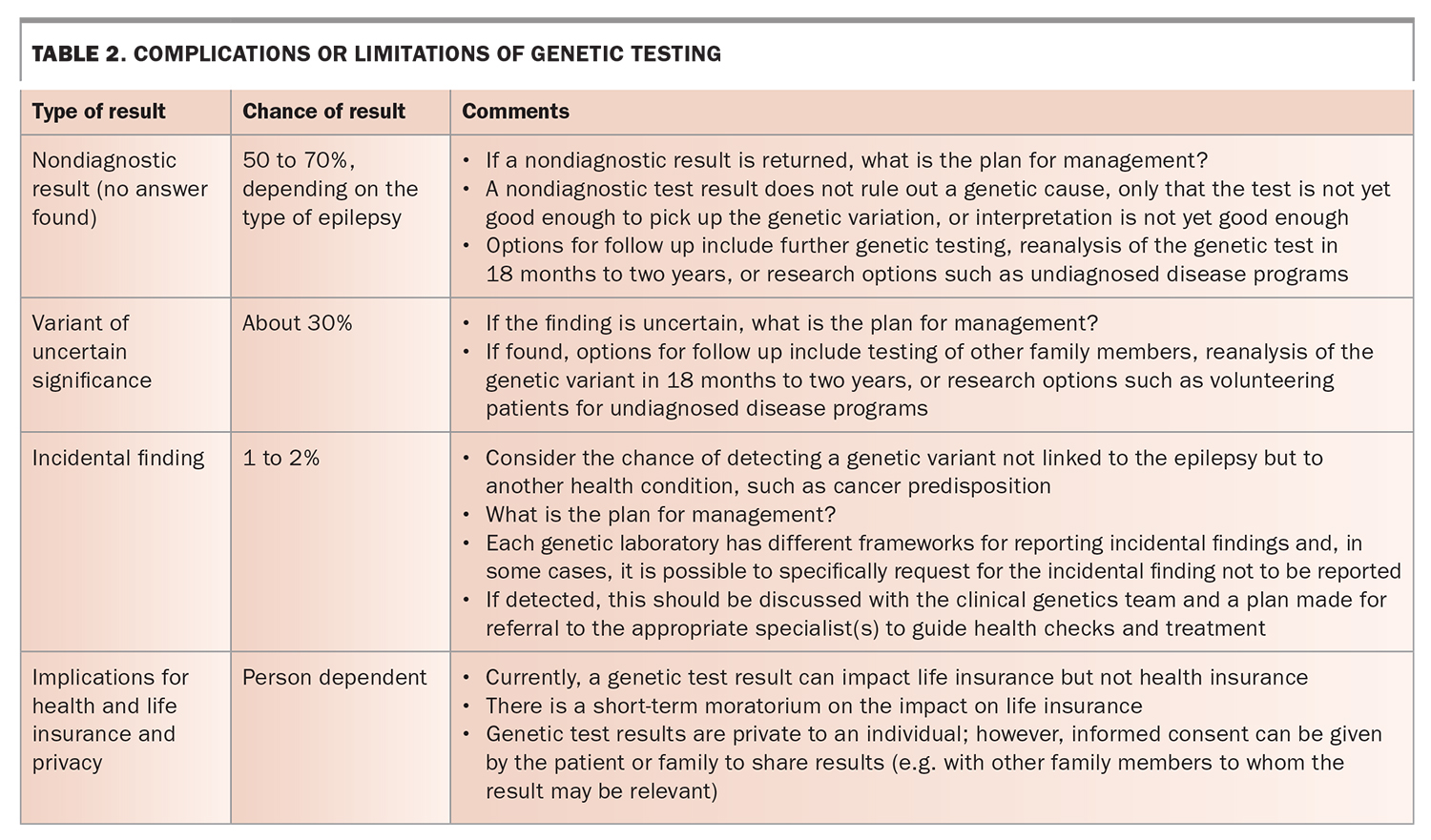
- The potential downsides or limitations of genetic testing (Table 2).
- Practical considerations. These include whether the test would receive a Medicare rebate, any out-of-pocket costs, what samples are required, the waiting time for results, who can order the testing and which referrals are required.
{kind=link}
Which genes are linked to epilepsy?
Over 600 genetic causes are currently linked to epilepsy.10 The genetic causes of DEEs reflect the wide range of genes involved in our most complex organ: the brain. About half of our 20,000 genes are thought to have a role in neurological development or function.
Almost weekly, new gene–epilepsy relationships are revealed.10,31 National and international consortia dynamically curate the genetic epilepsy landscape. For example, PanelApp Australia is a crowd sourced tool that takes a ‘citizen science’ approach to data collection, allowing gene panels to be shared, downloaded, viewed and evaluated by the scientific community (https://panelapp.agha.umccr.org). Experts interested in genetic epilepsy can nominate new genetic epilepsy genes and volunteer to review the evidence for such relationships.32 As of the 19th June 2022, there were 641 genes linked to genetic epilepsy (https://panelapp.agha.umccr.org/panels/202/).
What types of genomic changes can lead to epilepsy?
Changes in DNA within the nucleus of cells (nuclear DNA) that can cause a clinical phenotype such as epilepsy are traditionally separated into sequence variants and structural events.
- Sequence variants are small, single base pair substitutions or insertion-deletion (indel) events up to 50 base pairs in size. An example is a missense change in a gene that causes a change in the structure of the protein made by that gene.
- Structural events are variants of more than 50 base pairs in size and include copy number variants, deletions, duplications or much more complex changes in chromosomal structure such as inversions and translocations, where parts of chromosomes are crossed over or swapped between different chromosomes.
- Another type of genetic change that can cause certain epilepsies are repeat expansions. This is when a small region of DNA that is normally repeated expands, usually damaging the cell. An example is expansion of the repeat region at the start of the FMR1 gene, resulting in Fragile X syndrome.
- An array of changes affecting the DNA within mitochondria are now also recognised as causes of epilepsies, as well as changes in the epigenome, impacting the control of gene expression.18
Approaches to genetic testing for epilepsy
Although an array of genomic variation can cause genetic epilepsy, so far, we do not have one perfect genomic test to accurately and easily detect all these genomic possibilities. More complex genomic variants, such as noncoding variants, are particularly hard to detect and interpret. Therefore, a step-wise approach, starting with assessing for syndromic features, is considered the current benchmark for genomic testing (Flowchart 1).9
A genetic epilepsy diagnosis: what next?
A diagnosis is only the start of the journey for most patients and families, and the GP is ideally placed to translate a diagnosis into improved care and support. Box 4 highlights important aspects of care and support for patients and their family after a diagnosis, including links to helpful resources.
No genetic epilepsy diagnosis yet: what next?
Due to the limitations of current genetic testing and knowledge of the underlying causes of genetic epilepsy, 50 to 70% of patients undergoing testing will not receive a definitive genetic diagnosis.9,10 The lack of a diagnosis adds to uncertainty and impacts mental health, thus management needs to focus on symptom management, with recognition of the need for potential additional mental health and wellbeing support.33 The GP is ideally positioned to support such patients and their families in an integrated manner. In discussion with the involved neurologist and clinical genetics teams, they may consider other approaches, including different type of tests, reanalysis, or participation in research such as entry into a genetic epilepsy research program.25,34 Peer support is available for patients with a suspected genetic cause without a diagnosis and include Genetic Epilepsy Team Australia, which runs annual family conferences (https://www.geneticepilepsyteam.com.au/), and Syndromes Without A Name Australia (https://swanaus.org.au/).
Conclusion
Breakthroughs in genetic and epilepsy research are helping patients and their families better understand the cause of their underlying epilepsy. The decision to have a genetic test needs careful consideration and support from the patient, their family and any treating clinicians to ensure shared decision making, patient empowerment and improved health outcomes. Global research efforts continue to provide more rapid diagnoses, as well as better support and treatments to more families. The GP plays a crucial role in ensuring patients and their families benefit from integrated, person-centred and precise genetic epilepsy care. MT
COMPETING INTERESTS: None.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.