An acute generalised blistering eruption
Test your diagnostic skills in our regular dermatology quiz. What is the cause of this gradually progressive skin eruption in an otherwise healthy 54-year-old woman?
Case presentation
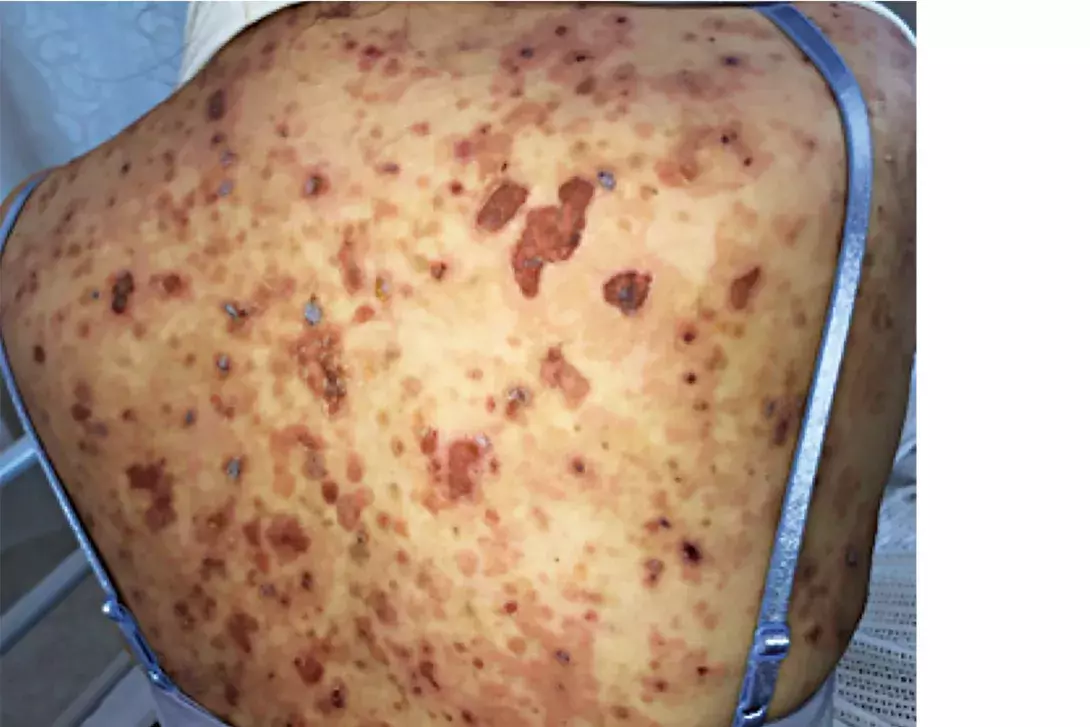
A 54-year-old woman presents with a one-month history of a gradually progressive, blistering eruption involving her trunk, arms and legs (Figures 1a and b). She has not started any new medications and has no other medical conditions.
Differential diagnoses
Conditions to consider among the differential diagnoses for an acute blistering eruption include the following.
- Erythema multiforme (EM). This acute self-limiting mucocutaneous eruption most commonly occurs as a result of infection, with herpes simplex virus (HSV) being the most common causative organism.1 Patients with EM minor are usually well; prodromal fever, myalgia and malaise are seen in EM major, a more severe clinical entity of EM that has far more extensive mucosal involvement.1 EM initially presents with pruritic and painful urticarial plaques that then develop into the characteristic erythematous concentric targetoid lesions that affect the distal extremities and favour extensor surfaces. The lesions show a large degree of morphological variability, with some atypical EM lesions presenting as haemorrhagic plaques that subsequently develop into blisters with crusting.1 Biopsy results are dependent on the evolutionary stage of the lesion, with epidermal necrosis a prominent finding in EM bullous lesions.1
- Bullous pemphigoid (BP). This autoimmune disease presents initially with pruritic urticarial plaques affecting the trunk and limbs that evolve into tense blisters.2 BP is the most common autoimmune vesiculobullous disease and most commonly affects individuals in the seventh decade of life and over.2 Disease is caused by circulating antibodies to bullous pemphigoid antigen 2 (BP180), an adhesional structure inherent to hemidesmosomes that tether basal keratinocytes to the basement membrane, resulting in subepidermal blisters.2 Biopsy is diagnostic and shows subepidermal blistering with a neutrophilic infiltrate; perilesional direct immunofluorescence shows linear deposition of IgG and complement C3 along the basement membrane.2
- Bullous viral exanthem. HSV, coxsackievirus, varicella-zoster virus, echovirus, enterovirus and adenovirus can result in vesiculobullous eruptions, which are commonly accompanied by systemic signs such as fever and lymphadenopathy.3 Eczema herpeticum occurs as a result of disseminated HSV-1 infection, resulting in vesiculopustular lesions on an erythematous base in an unwell patient with coexisting skin disease or immunosuppression.3 Coxsackieviruses A4 and A9 result in an acute onset of yellow vesicular crops and blisters affecting the face and trunk with accompanying fever.3 Varicella should always be suspected in an adult with a widespread, progressive vesiculobullous eruption in varying stages of evolution because of the increased risk of varicella hepatitis, encephalitis and pneumonitis.3
- Bullous impetigo. This is an acute blistering disease caused by exfoliative toxins (ETs) from Staphylococcus aureus infection. Local production of ETs at the site of infection results in acute, erosive, crusted lesions on an erythematous base affecting the face, torso, axillae and extremities in a patient who is otherwise well.4 Immunosuppressed individuals, such as patients with end-stage renal failure, are at risk of staphylococcal scalded skin syndrome, in which systemic dissemination of ETs cause bullous eruptions distal to the site of infection, and has a mortality rate of 60% in adults.4 ET-A and ET-B cause proteolysis of desmoglein-1, a desmosomal glycoprotein involved in intercellular adhesion, affecting keratinocytes in the stratum granulosum.4 Patients are highly contagious because of the subcorneal proliferation of S. aureus. Histopathological features of bullous impetigo include subcorneal blisters with an accompanying neutrophilic infiltrate in the superficial epidermis, while Gram staining shows Gram-positive cocci.4
- Pemphigus foliaceous. This is the correct diagnosis. Pemphigus foliaceous (PF) is a rare autoimmune disease affecting the superficial epidermal layers, in which autoantibodies to desmoglein-1 results in subcorneal acantholysis.5 PF can affect individuals of all ages, although it most commonly occurs in the sixth and seventh decades of life.5 Sporadic and endemic forms of PF exist – the latter most commonly occurs in rural Brazil, where it is known as Fogo Selvagem.5 Penicillins, ACE inhibitors, NSAIDs and barbiturates can also cause a drug-induced form of PF.5 PF presents acutely with painful, pruritic patches and erosions on an erythematous base affecting the head and trunk but spares the lower limbs.5 Depending on the extent of disease, these patients are usually well and have a positive Nikolsky sign, which describes induced detachment of the epidermis by mechanical pressure applied parallel to the skin surface (e.g. firmly pressing finger along the skin). Oral mucosal involvement in PF is rare, unlike the more serious form of this disease, pemphigus vulgaris.5 Pemphigus erythematosus is a clinical variant of PF that results in a facial butterfly-like distributed eruption affecting the malar skin, which may appear similar in morphology to that seen in systemic lupus erythematosus.5
Diagnosis
Biopsy is diagnostic for PF and will show acantholysis with an accompanying eosinophilic spongiosis, whereas direct immunofluorescence of perilesional skin shows IgG in the subcorneal epidermis.5 Serum autoantibodies to desmoglein-1 correlate with disease activity and are useful to diagnose PF and monitor response to therapy.5
Management
Pemphigus is a rare disease that tends to be chronic and difficult to treat. The treatment approach to PF has three phases: control (reduce number of new lesions), consolidation (achieve healing in 80% of lesions) and maintenance (prevent new lesions).5 The mainstay of treatment is oral corticosteroids, with other modalities such as pulsed methylprednisolone or plasmapheresis reserved for cases resistant to oral corticosteroids of 120 mg daily.5 Off-label use of adjuvant immunosuppressant medications such as azathioprine, cyclosporin, cyclophosphamide, methotrexate and mycophenolate may be used with systemic corticosteroids in refractory cases. Off-label use of rituximab has been described as being useful in halting disease progress.6 Topical emollients should be used to restore the skin barrier and prevent superimposed skin infections.
Outcome
This patient was admitted to hospital for investigation and management. A skin biopsy for histopathology and direct immunofluorescence was performed, which returned an acantholytic picture with intraepidermal clefting and positive intracellular IgG and C3. Pemphigus antibodies were detected by indirect immunofluorescence in blood. A pre-immunosuppression screen was performed with all parameters normal. She was diagnosed with PF and started on prednisone 50 mg daily, with rapid improvement. Initial treatment with rituximab resulted in a rapid improvement of her existing skin lesions. Treatment was subsequently changed and she remains on a slow taper of prednisone with azathioprine 100 mg daily as a corticosteroid sparer. MT