Systemic lupus erythematosus: reducing life-threatening progression
Lupus
Autoimmune diseases
Systemic lupus erythematosus is a multisystem autoimmune disease of unknown aetiology. It can affect almost any organ system and ranges from mild to severe to life-threatening.
- SLE can affect almost any organ system and ranges from mild to severe to life-threatening.
- There are no specific diagnostic criteria. Clinical criteria combined with immunological and or biopsy criteria are useful to make a diagnosis.
- The differential diagnosis is wide, and there is significant overlap with other rheumatological conditions that must be excluded.
- SLE may coexist with other autoimmune diseases such as rhupus syndrome, mixed connective tissue disease and overlap syndromes.
- SLE affects pregnancy outcomes and increases neonatal complications.
- Treatment intensity is directed by the severity and risk of organ involvement.
- Treatment with immunosuppression should be closely monitored for drug toxicity.
Systemic lupus erythematosus (SLE) is most prevalent in women (90% in most studies) and most common in the 14 to 64 years age range, suggesting a role for hormonal factors in its pathogenesis. There is also a genetic predisposition – SLE is more common in African, Asian and Indigenous Australian populations than Caucasian populations.1 The genetic predisposition is supported by a high prevalence among monozygotic twins and a higher prevalence in relatives of people with SLE.2
Characteristic features
SLE has protean clinical and laboratory manifestations. It has a variable course and prognosis, is marked by remission and relapses and ranges from mild to life-threatening. Immunological aberrations are manifested by excessive antibody production resulting in cytotoxic damage or immune complex formation resulting in inflammation.
Organ systems may be involved in isolation or in combinations. Involvement of vital organs such as the kidney and central nervous system account for the highest morbidity and mortality. Morbidity and mortality are a consequence of both the tissue damage from the disease process and/or the potent treatments required to control disease progression.1
Classification criteria
Clinical features range from constitutional to organ specific involving most organ systems including skin, mucous membrane, joints, kidney, brain, serous membranes, lung, heart and, occasionally, the gastrointestinal tract. Several of these clinical manifestations form part of the classification of SLE for research and study purposes and to help clinicians identify specific organ involvement and target treatment. However, sensitivity and specificity are limited and, although not diagnostic, these criteria are useful aids to clinicians for diagnostic purposes.
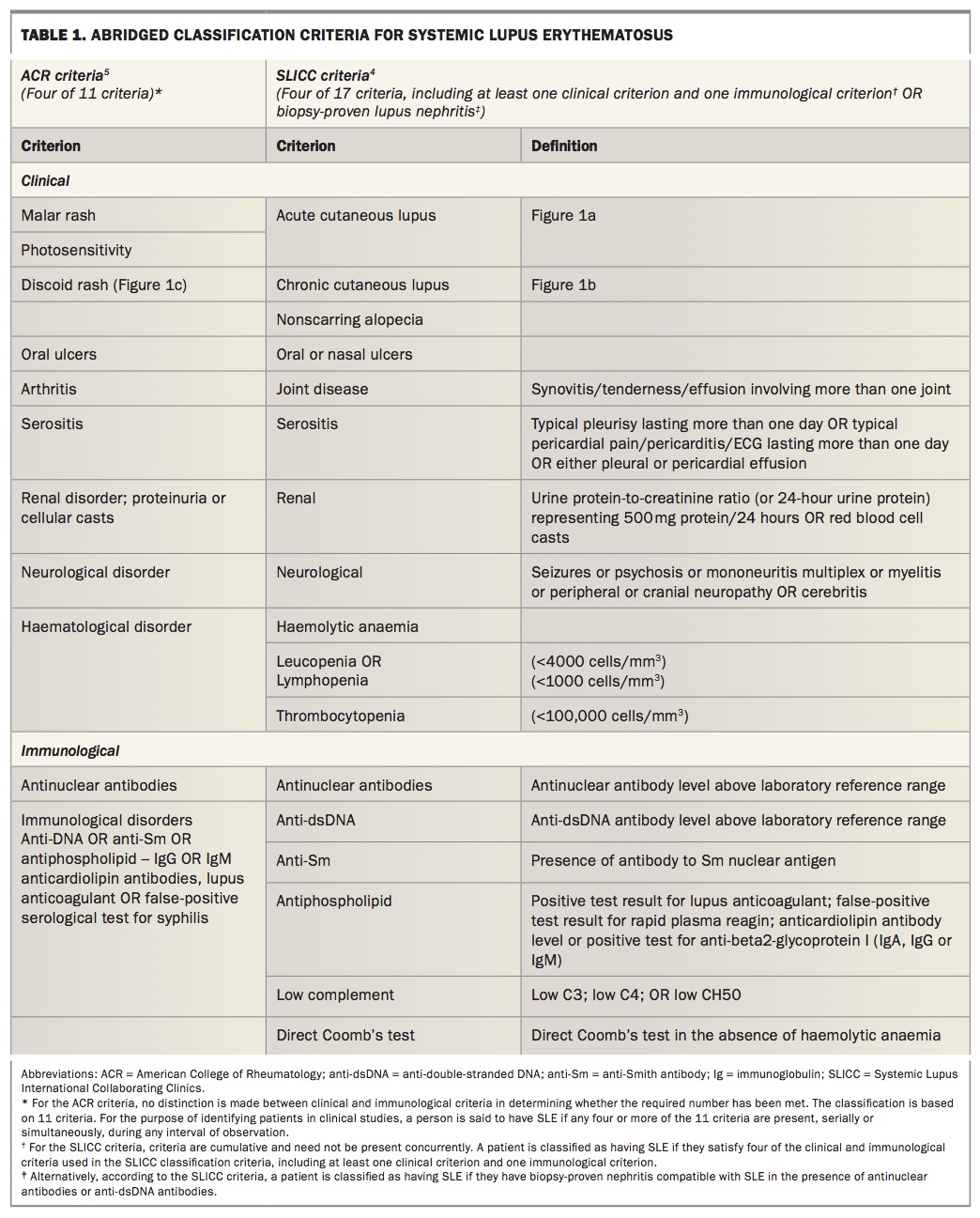
The 2012 Systemic Lupus International Collaborating Clinics (SLICC) – which is a revised version of the 1997 American College of Rheumatology (ACR) classification criteria – has enhanced the sensitivity of the criteria for patients with early disease and reduced the number of patients misclassified.3,4 This has however, come at the cost of reduced specificity. More recently there has been a push to develop newer criteria that maintain the sensitivity of the SLICC criteria and the specificity of the ACR criteria.
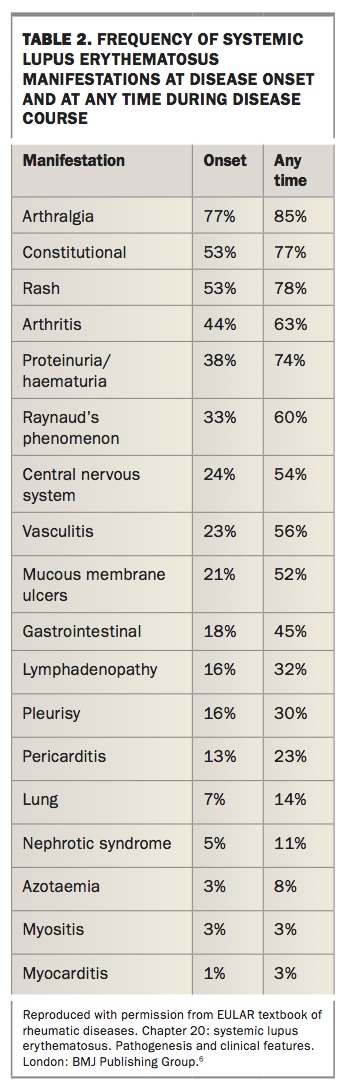
In the absence of diagnostic criteria, most rheumatologists use the classification criteria to aid in the diagnosis of SLE. However, classification criteria are not meant to be used as diagnostic criteria because they do not capture all aspects of the disease. It is therefore not unusual for a patient to not meet sufficient classification criteria but to still rouse enough clinical suspicion to be diagnosed and managed as SLE by the treating rheumatologist. Table 1 lists the classification criteria followed by a brief outline of the salient features of each manifestation.4,5 Table 2 outlines the frequency of different symptoms at disease onset and during the course of the disease.6
{kind=link}
{kind=link}
Constitutional symptoms
Fatigue, fever and weight loss are present in most patients with SLE, at some point in the course of their disease. These symptoms may be manifestations of active disease or complications of treatment. Myalgia is also common. Myositis is relatively uncommon.
Mucocutaneous
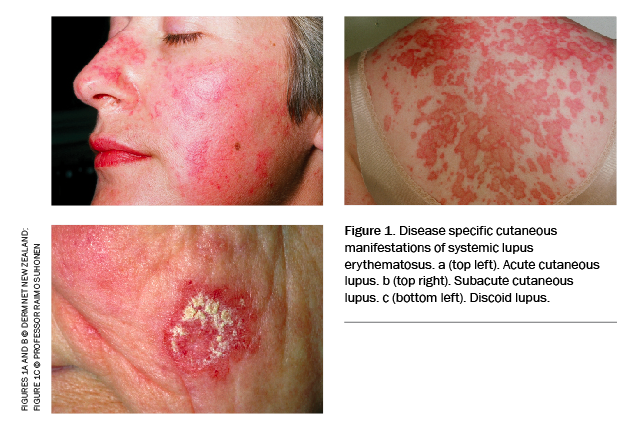
Skin and mucous membrane involvement and photosensitivity are common. The most common lesion – cutaneous lupus erythema (‘the butterfly rash’) – typically involves a malar distribution over the cheeks and nose but spares the nasolabial folds (Figure 1). It usually occurs after sun exposure. Patients may develop oral and/or nasal ulcers (typically painless), nonscarring alopecia or scarring alopecia (common in patients with discoid lupus erythematosus).7,8
{kind=link}
Musculoskeletal
Arthritis and arthralgia occur in more than 90% of patients (and may be the earliest manifestation) and may be migratory, polyarticular or symmetrical in nature. Although moderately painful, it does not usually cause bony erosions but can lead to fixed deformity – that is, Jaccoud’s arthropathy.9 ‘Rhupus syndrome’ is a relatively new clinical term. It denotes an erosive arthropathy with identical radiographic features to rheumatoid arthritis while also meeting the clinical criteria for SLE.
Cardiopulmonary
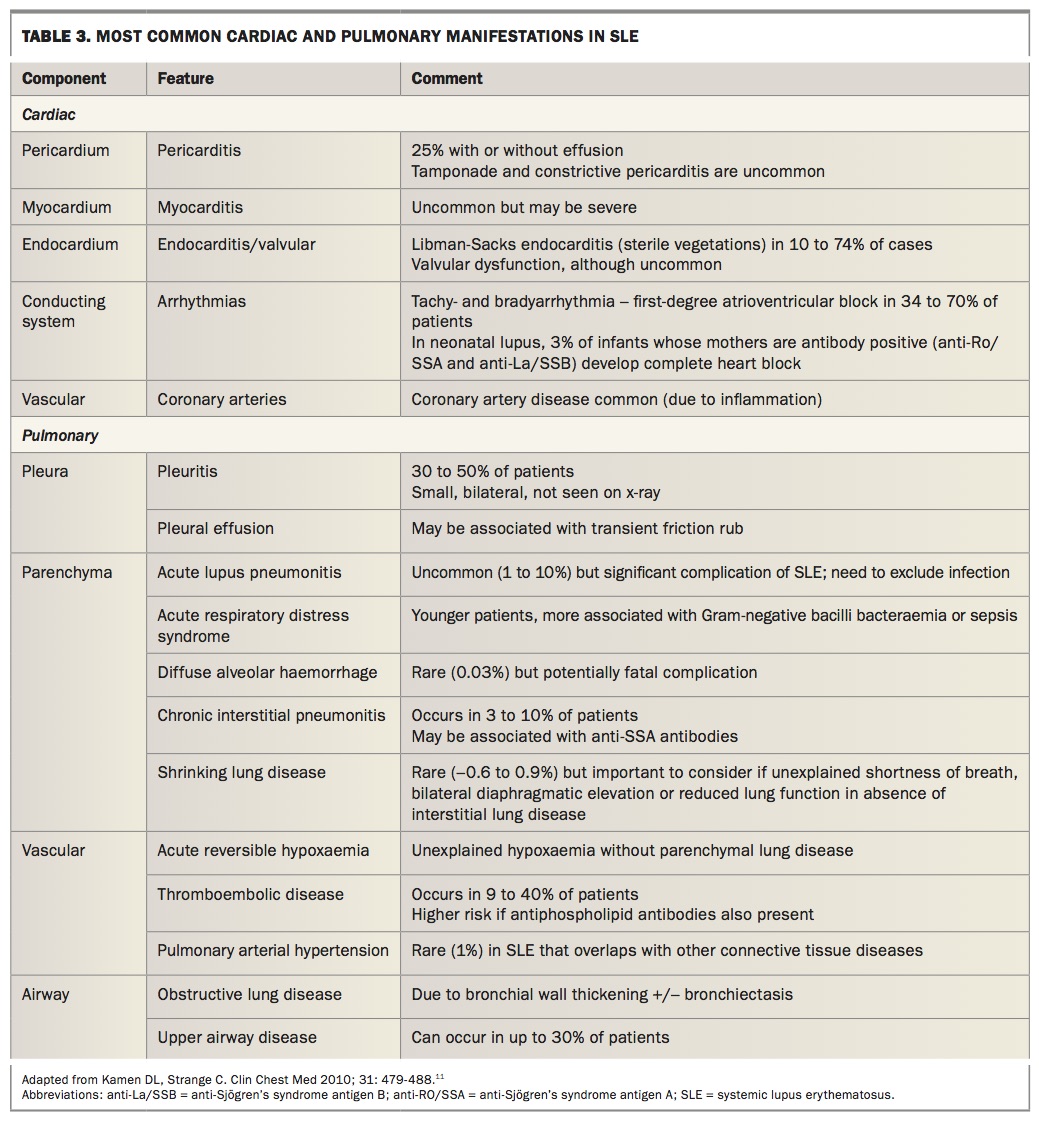
Cardiac disease is not uncommon in patients with SLE and includes the pericardium, myocardium, valves and conduction system. Premature coronary artery disease development is a grave concern in SLE, driven by chronic systemic inflammation and an increasing cause of premature death.10 Table 3 outlines the most common cardiac and pulmonary manifestations of SLE with clinical clues to diagnosis.11
{kind=link}
Renal
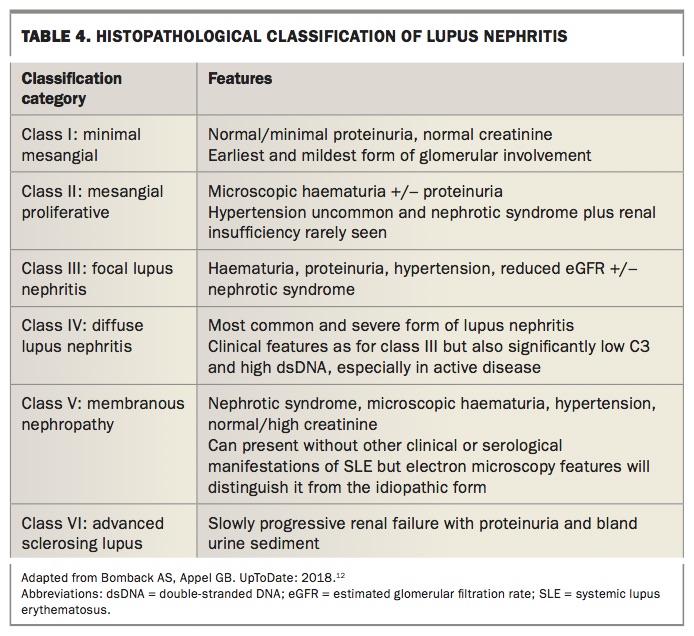
Renal disease occurs in 30 to 60% of patients, with the most common renal abnormality being proteinuria more than 0.5 g daily All patients with SLE should therefore undergo regular screening with urinalysis for red cells and cellular casts, urine protein-to-creatinine ratio on spot urine sample, creatinine and glomerular filtration rate. A renal biopsy is paramount in determining the severity of lupus nephritis, which guides therapy. Table 4 outlines the classification features of lupus nephritis.12,13
{kind=link}
Haematological
Neutropenia is thought to be due to increased peripheral destruction of neutrophils due to circulating antineutrophil antibodies, increased margination and decreased marrow production due to T cell or monocyte-mediated suppression of granulocytopoiesis.14 Lymphopenia is due to antilymphocyte antibodies or increased apoptosis. Thrombocytopenia is due to increased peripheral destruction due to antiplatelet antibodies. Impaired production and splenic sequestration may occur in some patients. Anaemia of chronic disease frequently develops during disease flares, whereas autoimmune haemolytic anaemia with reticulocytosis can occur due to warm-type IgG anti-erythrocyte antibodies.
Neurological
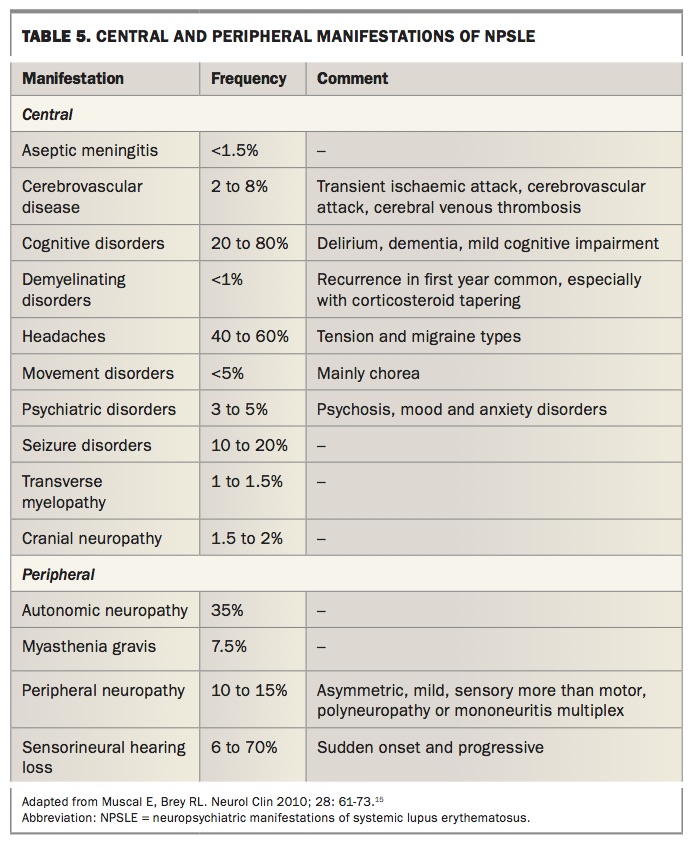
Neuropsychiatric manifestations of SLE (NPSLE) can occur in the absence of serological evidence or other SLE manifestations. The ACR has defined 19 central and peripheral manifestations of NPSLE, which are outlined in Table 5.15-19 It is important to remember that these manifestations can also occur in the absence of SLE.
{kind=link}
Gastrointestinal
Gastrointestinal symptoms occur in up to 40% of patients with SLE and most are due to adverse drug reactions and/or viral or bacterial infections. These include oesophagitis, intestinal pseudo-obstruction, protein losing enteropathy, lupus hepatitis, acute pancreatitis, mesenteric vasculitis or ischaemic peritonitis.
Ophthalmological involvement
Keratoconjunctivitis sicca is the most common manifestation of ophthalmological involvement and usually indicates development of secondary Sjögren’s syndrome. Less common are retinal vasculopathy, optic neuropathy, choroidopathy, scleritis, episcleritis and anterior uveitis (iritis, iridocyclitis).
Other associated conditions and complications
Immunodeficiencies
Although rare, a defect in C1-inhibitor is associated with inflammatory and autoimmune disorders including SLE. Similarly, patients with complete C4-inhibitor deficiency can present with SLE.
Antiphospholipid syndrome
This is associated with antiphospholipid antibodies, which can be detected in 40% of patients with SLE. The main manifestations are thromboembolic disease and poor obstetric outcomes, especially multiple spontaneous abortions, pre-eclampsia and premature birth.
Osteonecrosis
The estimated risk of osteonecrosis varies from 3 to 40%. It is thought to be related to avascular necrosis with the underlying disease as well as concomitant use of glucocorticoids.
Osteoporosis
Osteoporosis is a common complication of SLE and/or medications prescribed for its treatment.
Infection
Serious infection, especially of the skin and respiratory and urinary tracts, develops in 50% of patients with SLE, with the majority of infections due to pathogenic bacteria. However, opportunistic infections can occur due to fungi and are usually related to the intensity of immunosuppressive therapy. They are a common cause of mortality. Viral infections are more common, including infections from parvovirus B9 (which can cause a lupus-like syndrome), Epstein-Barr virus, cytomegalovirus, varicella zoster virus and human papilloma virus. Mycobacterial infections including nontuberculous have been noted more frequently in patients with SLE.
Other autoimmune diseases
There is an increased prevalence of thyroid disease and myasthenia gravis in patients with SLE. There is also a high prevalence of autoimmune conditions among relatives of patients with SLE.
Immunological markers of SLE
Diagnosis depends on a combination of clinical and immunological criteria. Despite the availability of nonoperator- dependent and automated assays, the immunofluorescence assay is still considered the gold standard for detecting autoantibodies to a wide range of nuclear and cytoplasmic antigens (antinuclear antibody [ANA] test).20 It should be noted that a positive ANA test, although sensitive, is not specific or diagnostic of SLE. Most patients with a positive ANA result will not have SLE. About 5% of patients with SLE are ANA negative by indirect immunofluorescence and relates to poor testing methods used to detect ANA.
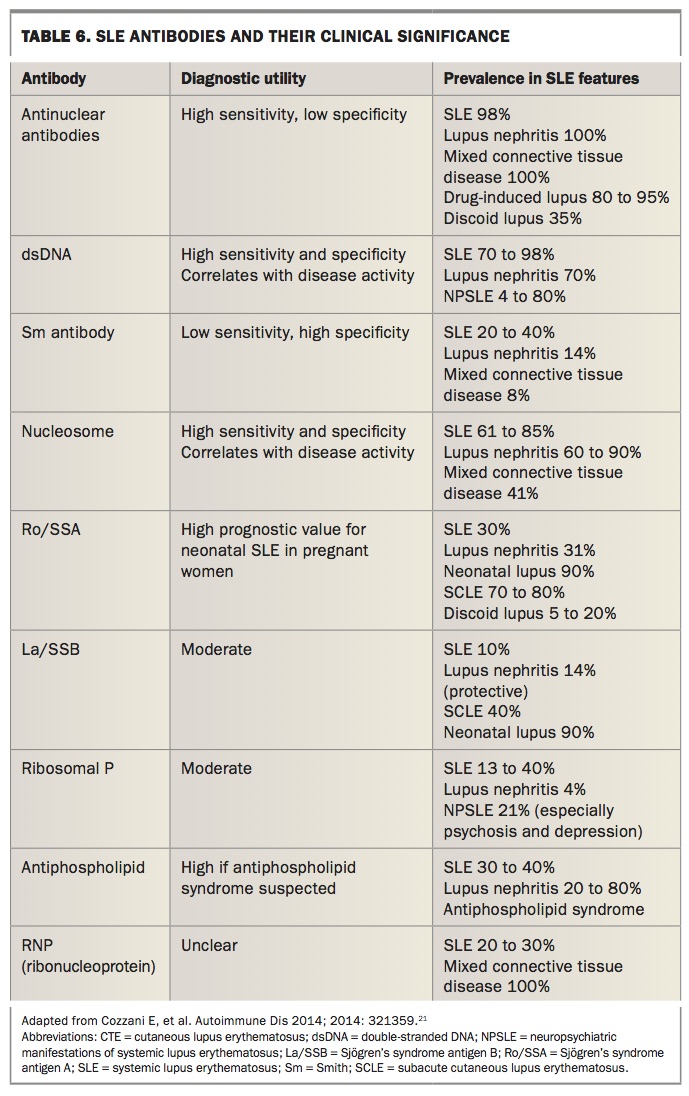
Anti-dsDNA-antibody testing is more useful diagnostically and can be done by Farr assay (40% sensitive; 90% specific) and/or enzyme-linked immunosorbent assay (ELISA; 90% sensitive; 70% specific). Unfortunately there is lack of standardisation for all auto antibody testing in laboratories. Table 6 outlines antibodies found in SLE and their significance in the diagnosis of SLE.21,22
{kind=link}
Diagnosis
A diagnosis of SLE is a clinical judgement based on the constellation of symptoms and signs in the setting of supportive serological studies after excluding alternative diagnoses. There is a great deal of variability in expression and severity, making a definite diagnosis of SLE difficult. The classification criterion is mainly designed for research purposes but clinicians can refer to this in terms of making a diagnosis of classic SLE. The differential diagnosis is wide, and there is significant overlap with other rheumatological conditions that must be excluded before SLE is diagnosed (Box 1).
{kind=link}
As a general guide the following applies after excluding alternative diagnoses:
- definite SLE – patients who fulfil the 1997 ACR criteria or 2012 SLICC criteria
- probable SLE – patients who do not fulfil the classification criteria for SLE but have two to three diagnostic criteria or those who have other SLE manifestations not included in the other classification criteria
- possible SLE – patients who only have one of the ACR/SLICC criteria in addition to at least one or two of the uncommon features
- undifferentiated connective tissue disease – even fewer features suggestive of SLE
- ANA-negative SLE – less than 5% of patients with SLE are negative for ANA.
Treatment
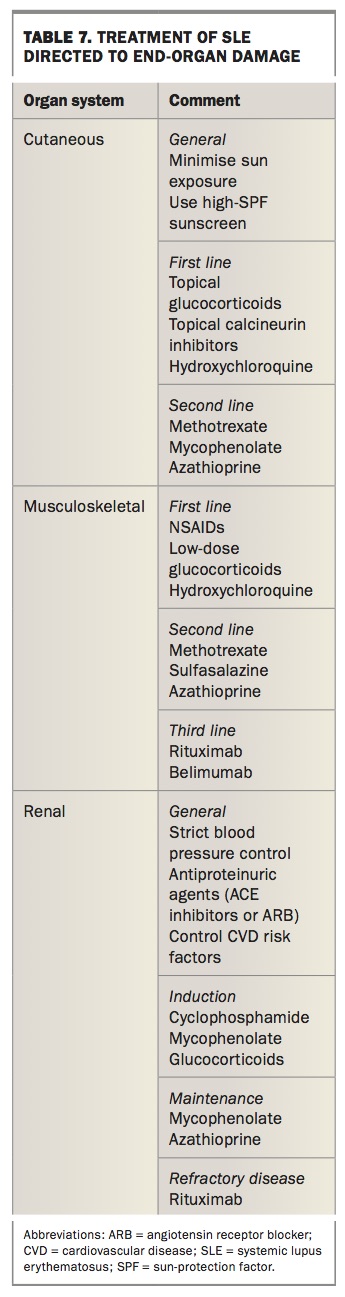
Treatment of SLE can broadly be divided into pharmacological and nonpharmacological approaches. Treatment is guided by specific end-organ involvement, includes remission induction followed by maintenance and aims to reduce organ failure and mortality (Table 7).
{kind=link}
Nonpharmacological
All patients should receive information about SLE at the time of diagnosis (from, for example, Arthritis Australia and Lupus Australia). Patients should be encouraged to maintain a healthy lifestyle incorporating a balanced diet and exercise program.23,24 An inverse relationship between vitamin D levels and SLE activity suggest that ensuring normal vitamin D levels is advisable.25,26 Smoking has been implicated in the pathogenesis of SLE, and patients should be encouraged to quit.27 A causative role of ultraviolet radiation is strongly associated with cutaneous lupus erythematosus and patients should be advised to minimise harmful exposure and to use sunscreen and other sun-protective measures.28,29
Pharmacological
The treatment options for the most common manifestations of SLE, namely cutaneous, musculoskeletal and renal will be discussed.
Cutaneous
High-potency corticosteroid creams have been shown to be significantly superior to low-potency corticosteroids. Prolonged use should be avoided due to known side effects (atrophy, telangiectasia).30 Topical calcineurin are as effective but with a better side effect profile than topical corticosteroids.30 However, recent studies raise concern regarding the risk of haematological and cutaneous malignancies with exposure to topical or oral calcineurin inhibitors, prompting a FDA black box warning.31
Patients who fail to respond to topical therapies are offered systemic agents. Initially, the antimalarial hydroxychloroquine up to 5 mg/kg daily (of actual body weight) to a maximum of 400 mg daily is offered. It is generally well tolerated, aside from occasional temporary gastrointestinal discomfort. A rare but serious side effect is the development of an irreversible retinopathy due to accumulation of hydroxychloroquine in the retina following prolonged use, especially beyond five years with doses of more than 6.5 mg/kg daily. Regular ophthalmological review biannually for the first five years after diagnosis and annually after five years is generally recommended.32 For recalcitrant cases, stronger immunosuppressive mediations such as methotrexate, mycophenolate and azathioprine can be prescribed.
Musculoskeletal
Despite the high prevalence, few data guide therapy of these manifestations. Although NSAIDs can be used to treat flares of arthritis in SLE, hydroxychloroquine is the preferred option for long-term management.33 Low-dose glucocorticoids (up to 15 mg daily) are also frequently used to control flares of SLE arthritis. However, due to their significant side effect profile, the duration is kept as short as possible.
Those with refractory symptoms are offered more potent immunosuppressive therapy. Methotrexate has been shown in controlled trials to reduce symptoms and have a corticosteroid-sparing effect in this cohort.34 Although generally a safe medication when taken as prescribed (weekly dosing of 10 to 25 mg plus folic acid), the main side effects include gastrointestinal and stomatitis (common) and deranged liver function tests or bone marrow suppression (uncommon). Women of reproductive age should be educated on contraception and potential teratogenic risk to pregnancy; men should be advised to use contraceptive measures when taking methotrexate. In those intolerant of or unresponsive to methotrexate, leflunomide has limited data for benefit. Its toxicity profile and precautions are similar to methotrexate.35 Methotrexate and leflunomide require regular blood test monitoring for safety (blood count and liver function tests monthly initially, followed by three monthly once stable).
Other disease-modifying drugs such as azathioprine, mycophenolate and cyclophosphamide have been used extensively in SLE nephritis but high-quality evidence of their role in SLE arthritis is lacking. Biological agents that are effective in other rheumatological conditions have not been beneficial in SLE and some, for example the tumour necrosis factor blockers, may actually induce SLE.
Although B lymphocytes are key mediators of disease in SLE, B cell depletion via rituximab has been disappointing in clinical trials. But it has found a niche in treatment-resistant SLE.36 Belimumab, a human monoclonal antibody inhibitor of B cell activating factor (BAFF), has shown promising benefit as a safe and well-tolerated drug for musculoskeletal manifestations.37,38
Renal
Lupus nephritis (LN) is one of the most serious complications of SLE and a leading cause of morbidity and mortality. The aims of treatment are induction and maintenance of remission while preventing progression to end-stage renal disease and minimising drug-related toxicity.
Mild disease (biopsy class I/II) is treated with supportive measures and other disease-modifying drugs such as azathioprine and mycophenolate. Class III/IV indicates proliferative LN has a worse renal prognosis and is usually the target of trials and treatment. Glucocorticoids are efficacious in rapidly controlling active disease and have been the standard of care until landmark trials showed better outcomes with combination of pulsed cyclophosphamide.39 Cyclophosphamide, however, has a significant side effect profile with increased risk of infection, haematological toxicity, bladder malignancy, teratogenicity and impaired fertility including risk of ovarian failure, which is particularly relevant for this cohort that largely comprises young women. The Euro-Lupus Nephritis Trial successfully showed that a lower dose of cyclophosphamide regimen had similar efficacy but lower toxicity when used in patients with milder LN.
Mycophenolate, an oral agent that inhibits lymphocyte proliferation and antibody formation by inhibiting the enzyme inosine monophosphate dehydrogenase, has emerged as an alternative to cyclophosphamide as a remission-inducing agent, with high-quality data showing equal efficacy with a better side effect profile, especially in regards to ovarian failure.39 Gastrointestinal, haematological and infections are the most common side effects seen with mycophenolate.
Studies on biological agents for LN have been disappointing despite earlier promising results.39 The belimumab studies were not designed for LN, as patients with severe LN were excluded. However, a pooled post hoc analysis showed improvement in renal parameters.40
Maintenance immunosuppression is required to minimise risk of relapse and disease progression. Azathioprine and mycophenolate have demonstrated similar efficacy as cyclophosphamide, with a better safety profile in maintaining remission in patients with LN.41 Azathioprine reduces the number of circulating B lymphocytes and T lymphocytes by interfering with purine synthesis. Gastrointestinal issues, alopecia and stomatitis are common side effects. Dose-related myelosuppression is an uncommon but serious adverse effect that requires close monitoring and thiopurine methyltransferase activity testing before initiation. Significant dose reduction is required in those taking xanthine oxidase inhibitors (e.g. allopurinol for gout).
Monitoring patients with SLE
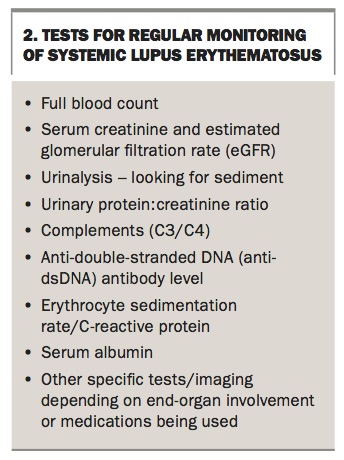
There are no specific or consistent guidelines for the monitoring of patients with SLE.42 However, as a rule, patients with active disease or end-organ damage are managed more frequently or for specific indications. For patients with mild or inactive disease, annual assessment is reasonable. In those with active disease, at least three to six monthly monitoring would be advisable. Additional or earlier assessment is warranted for patients planning pregnancy, surgery, change in medication or development of new significant symptoms or organ involvement. Investigations will depend on the individual patient and end-organ involvement. The most useful tests for regular monitoring are listed in Box 2.
{kind=link}
Conclusion
Initial evaluation for SLE requires a careful history and physical examination, along with selective laboratory testing to identify features that are characteristic of SLE or that suggest an alternative diagnosis. Baseline assessment includes complete blood count and differential as well as serum creatinine level and urinalysis in all patients suspected of SLE. Diagnostic imaging or biopsy, depending on the organ involved, should also be considered. Treatment is targeted to prevent end-organ damage, morbidity and mortality. Careful monitoring of patients with SLE for disease activity, end-organ damage, development of comorbidities and drug toxicity is important for optimal management of this disease. MT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.