Crystal arthritis – managing gout and calcium pyrophosphate deposition disease
Dr Baquir is an Advanced Trainee in the Department of Rheumatology, Westmead Hospital, Sydney.
Dr Yoon is an Advanced Trainee in the Department of Rheumatology, Westmead Hospital, Sydney.
Dr Cai is a Staff Specialist Rheumatologist in the Department of Rheumatology, Westmead Hospital, Sydney; the Clinical Lead of the Osteoarthritis Chronic Care Program for Western Sydney Local Health District; and a Clinical Senior Lecturer for The University of Sydney, Sydney, NSW.
SERIES EDITOR: Associate Professor Arvin Damodaran BSc, MB BS(Hons), MMedEd, FRACP, representing the Education Training and Workforce Committee of the Australian Rheumatology Association.

Gout
Arthritis
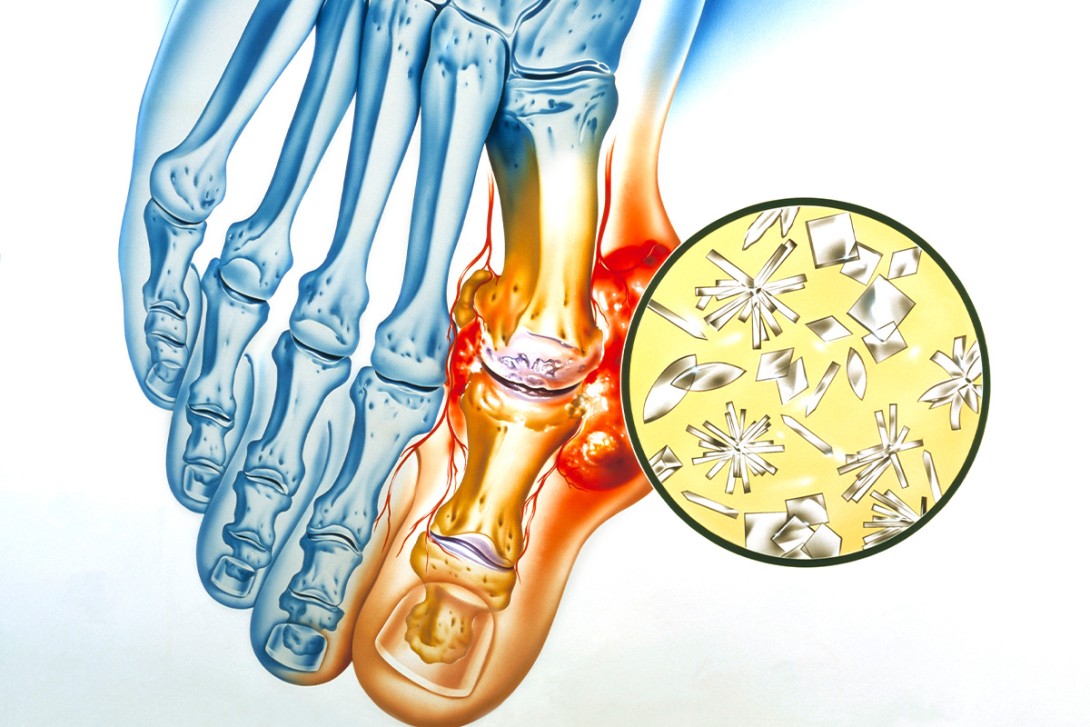
Crystal arthritis is one of the most commonly encountered musculoskeletal presentations in primary care. Most of these presentations are related to gout or calcium pyrophosphate deposition disease (CPPD), with patients treated for acute and chronic arthritis. Anti-inflammatory therapies are available for the management of acute gout flares and CPPD, and urate-lowering therapy for long-term gout management.
Correction
A correction for this article appears in the May 2024 issue of Medicine Today and available here. The online version and the full text PDF of this article have been corrected.
- Crystal arthritis, the deposition of crystals in the joints and soft tissues, typically present as gout and, less commonly, calcium pyrophosphate deposition disease (CPPD).
- CPPD has multiple phenotypes, including acute CPP crystal arthritis (formerly known as pseudogout), chronic CPP crystal inflammatory arthritis, osteoarthritis with CPPD and asymptomatic chondrocalcinosis.
- Demonstration of crystals in synovial fluid on polarising light microscopy is the gold standard for diagnosis but clinical features and imaging can assist.
- Although pharmacotherapies are available to manage crystal arthritis, treatment remains suboptimal in Australia.
- Patient education and urate-lowering therapy (ULT) following a treat-to-target serum urate approach are essential in the management of gout. Flare prophylaxis is recommended in patients commencing ULT.
- There are no disease modifying therapies for CPPD and anti-inflammatories are the current mainstays of treatment.
Crystal arthritis is one of the most commonly encountered musculoskeletal presentations in primary care. It usually presents as an acute monoarthritis caused by the deposition of monosodium urate (MSU) monohydrate (gout) or calcium pyrophosphate (CPP) dihydrate crystals (acute CPP crystal arthritis, previously referred to as ‘pseudogout’) in articular and nonarticular structures. Basic calcium phosphate (hydroxyapatite) crystals have also been implicated in conditions including osteoarthritis, Milwaukee shoulder syndrome, calcific periarthritis and tendonitis. In this article, we review the clinical, diagnostic and management aspects of gout and calcium pyrophosphate deposition disease (CPPD).
Gout
Epidemiology of gout
In Australia, the community prevalence of gout in adults ranges between 2 and 7%.1-4 The prevalence increases with age, is higher in men than in women and varies geographically and between ethnicities, with Māori and Pacific Islander people of New Zealand having the highest prevalence of gout globally.2,5,6 People of Māori and Pacific Islander background living in Australia are disproportionately affected by gout and have higher rates of hospitalisation and emergency department presentations.7 In addition to pain and disability, qualitative studies have shown the substantial social impacts of gout on patients and their families.8,9 In 2019-20, gout-related health expenditure in Australia was almost $234 million, with hospital-related services accounting for 55% of the total cost.10 Gout is also associated with other medical conditions, including hypertension, dyslipidaemia, cardiovascular disease (CVD), diabetes, chronic kidney disease and obesity.11
Despite effective treatments for gout being readily available, the condition continues to be undertreated. Australian studies have found that only 50% of people with gout were prescribed urate-lowering therapy (ULT) and adherence to therapy was suboptimal.3,4 Because of the profound disability, social impact and costs related to gout management, effective treatment at the primary care level remains the cornerstone of managing this condition.
Clinical presentation of gout
Gout presents as an intensely painful acute inflammatory arthritis (gout flare), usually involving a lower limb joint. The involvement of the first metatarsophalangeal joint (podagra) is most characteristic but there also can be involvement of the foot, ankle and knee as well as elbows, wrists, hands and periarticular structures (bursae, tendons, entheses). Upper limb involvement usually occurs in patients with longstanding and poorly controlled disease. Gout flares typically present as a monoarthritis; however, oligoarticular and polyarticular flares can occur in patients with poorly controlled disease which may also be accompanied by systemic symptoms such as fever.
The pain associated with gout can be described as stabbing, gnawing, burning or throbbing and reaches peak intensity in a short period of time (typically less than 12 hours from symptom onset). This is accompanied by clinical features of inflammation, including variable swelling, warmth and erythema at the affected site. The intensity of the pain can be severe, with marked limitation in function, difficulty walking and fear of even minimal contact. Without treatment, the flare typically settles within one to two weeks, followed by a pain-free asymptomatic period until another gout flare occurs. Most patients have recurrent flares that can lead to the development of tophi, chronic gouty arthritis and structural joint damage; however, some patients may have a single gout flare without any further symptoms. Tophi present as subcutaneous nodules under the skin, typically occurring at the joints (Figure 1a), ears, olecranon bursae, finger pads, tendons (e.g. Achilles), often with overlying vascularity, and can vary in size and may discharge ‘chalk-like’ material. Joint deformity and damage are common in patients with tophaceous gout and, in the event of ulceration, can be complicated by superimposed infection.

Risk factors for gout
The most important risk factor for gout is hyperuricaemia, with a concentration-dependent relationship between serum urate levels and the risk of developing gout.12 Factors that contribute to hyperuricaemia include metabolic syndrome, medication use (particularly diuretics and ciclosporin) and chronic kidney disease.13 Diet appears to have little effect on serum urate levels.14 However, high dietary intake of purine-rich foods, such as seafood, red meat, alcohol (beer and spirits) and sugar-sweetened beverages (high in fructose corn syrup) are known to increase the risk of gout flares.15-17 Urate metabolism and excretion also differ between ethnicities, which may account for the high incidence of gout in people of Māori and Pacific Islander background.18 Evidence on the risk of gout in Aboriginal and Torres Strait Islander people is conflicting, with recent cohort studies challenging earlier data that suggested a lower risk compared with the non-Indigenous population.19-21
Pathophysiology of gout
The development of gout starts with hyperuricaemia, which is predominantly caused by a renal underexcretion of uric acid rather than overproduction. However, it is important to recognise not all patients with hyperuricaemia develop gout. In people with severe hyperuricaemia (serum urate level ≥0.60 mmol/L), less than half developed gout over the next 15 years.22 The formation of MSU crystals occurs in some patients with hyperuricaemia and appears to be dependent on serum urate concentration or local joint factors, such as temperature, pH and concentration of sodium ions.23 Gout flares occur when MSU crystals activate the NLRP3 inflammasome, triggering a cascade of other proinflammatory cytokines and chemokines. Advanced gout is characterised by chronic synovitis, joint damage and tophi, which represents a chronic, granulomatous response to MSU crystals.24
Diagnosis of gout
Although a diagnosis can be made on clinical and radiographic findings, polarised light microscopy remains the gold standard for diagnosing gout.13 MSU crystals in synovial fluid from a symptomatic joint or tophi appear as needle-shaped and are negatively birefringent under polarising light microscopy. The detection of MSU crystals can exclude the presence of infection (e.g. septic arthritis, cellulitis, septic bursitis, osteomyelitis), which is the most important differential diagnosis for gout.
Blood tests
In the presence of typical symptoms and signs (e.g. podagra), gout can be diagnosed with a high level of confidence without the need for joint aspiration.25 Importantly, serum urate concentration is often normal during an acute gout flare and the level should be rechecked two to four weeks after resolution of the flare.26 Furthermore, hyperuricaemia alone is not sufficient to make a diagnosis of gout.
Imaging
If there is diagnostic uncertainty and when joint aspiration is unavailable or unfeasible, imaging, including ultrasonography and dual-energy CT (DECT) where available (Figure 1b), may aid the diagnosis. Conventional radiography (x-rays) may be used to determine structural damage in patients with advanced gout (Figure 1c). Findings of gout on ultrasound include a double contour sign (reflecting MSU crystal deposition on the surface of hyaline articular cartilage), tophi and aggregates.27-29 Of note, the absence of MSU deposits on ultrasound or DECT does not exclude a diagnosis of gout, particularly in the early stages of disease.


Management of gout
All patients diagnosed with gout should receive education about the condition. Modifiable risk factors for hyperuricaemia (e.g. metabolic syndrome) should be addressed and screening performed for associated comorbidities, including hypertension, dyslipidaemia, CVD, diabetes and chronic kidney disease. Less emphasis should be placed on dietary modification and weight loss, as these have only been shown to have a modest effect on reducing serum urate levels, and most people with gout will not achieve their target serum urate level with dietary modification alone.30
Pharmacological treatment can be divided into acute gout flare management and long-term gout management (i.e. ULT; Table 1). Lack of patient adherence to ULT remains the greatest barrier to effective treatment.

Anti-inflammatory therapy for acute gout flare management
Early administration of anti-inflammatory therapy is recommended to reduce joint pain and inflammation. The choice of anti-inflammatory medication should consider the time since onset of the gout flare, the number and size of joints affected, patient comorbidities and previous patient experiences with anti-inflammatory medications. Options include oral colchicine, NSAIDs or corticosteroids (oral, intra-articular or intramuscular; Table 1). Importantly, during a gout flare, patients should not discontinue ULT if they are taking it, and health professionals should discuss the benefits of ULT in managing the underlying cause of disease and preventing future flares and joint damage with the patient. Nonpharmacological therapies include the use of ice packs, taking rest and maintaining adequate hydration.31
Urate-lowering therapy for chronic gout management
The long-term management of gout involves continuous ULT prescribed at a dose that achieves MSU crystal dissolution. Management should follow a treat-to-target approach, and for most people with gout the recommended target serum urate level should be less than 0.36 mmol/L. This target may be even lower (<0.30 mmol/L) in people with severe gout (tophi, frequent flares, chronic gouty arthritis). ULT is not indicated for people with asymptomatic hyperuricaemia.32,33 Patients initiating ULT often experience gout flares, therefore, flare prophylaxis should be considered in all patients commencing ULT.
When to initiate urate-lowering therapy
ULT is strongly recommended for patients with gout who have frequent flares (defined as two or more flares per year), tophi or radiographic evidence of joint damage. ULT is not required for patients who have had their first gout flare, but should be considered if these patients have marked hyperuricaemia (serum urate level ≥0.54 mmol/L), uric acid nephrolithiasis or chronic kidney disease (stage 3 or greater).33 Allopurinol can be initiated during a gout flare, provided concurrent anti-inflammatory prophylaxis has also been commenced. This approach may improve adherence to ULT.34,35 Once ULT has been commenced, it should be continued lifelong. The cessation of ULT can also precipitate a gout flare.
Urate-lowering therapy options
Allopurinol, a purine-based xanthine oxidase inhibitor, is the preferred first-line option for ULT. It is generally well tolerated but common side effects include gastrointestinal upset (nausea, vomiting, diarrhoea) and rash.13 Rarely, it can cause severe cutaneous reactions that are strongly associated with the HLA-B*5801 allele, which is often seen in Southeast Asian (Han Chinese, Korean, Thai) and African American populations.36 Where available, genetic testing is recommended for at-risk individuals and, if the HLA-B*5801 allele is present, allopurinol is contraindicated.33 Allopurinol should be started at a low dose and gradually increased until the target serum urate is achieved:32,33
- for patients with normal renal function – initially 100 mg daily and increased by 100 mg increments every month
- for patients with an estimated glomerular filtration rate (eGFR) below 60 mL/min/1.73m2 – initially 50 mg daily and increased by 50 mg increments every month.
The average allopurinol dose required to reach serum urate concentrations below 0.36 mmol/L is about 400 mg daily and the maximum dose approved in Australia is 900 mg daily.37 Allopurinol is PBS listed for chronic gout.
Febuxostat, a nonpurine xanthine oxidase inhibitor, is the second-line option for ULT in people with gout who are unable to tolerate allopurinol.33 The initial dose of febuxostat is 40 mg daily, gradually increased by 40 mg increments every month until the target serum urate is achieved. The maximum dose approved in Australia is 80 mg daily. Febuxostat is PBS listed for chronic gout in patient with medical contraindication, documented hypersensitivity or intolerance to allopurinol. Recently, two large randomised controlled trials, CARES (Cardiovascular Safety of Febuxostat and Allopurinol in Patients with Gout and Cardiovascular Morbidities) and FAST (Febuxostat versus Allopurinol Streamlined Trial), examined the relationship between xanthine oxidase inhibitors and CVD.37,38 The CARES study found febuxostat was associated with a higher risk of all-cause and cardiovascular mortality compared with allopurinol in people with established CVD. This led to the recommendation that febuxostat should be avoided in patients with prior CVD. However, the CARES trial was criticised for its study design and high drop-out rate, with only 45% of patients completing all trial visits. In contrast, the FAST study found no difference in CVD outcomes between allopurinol and febuxostat. However, only 33% of the participants in the FAST study had a prior history of CVD and the study excluded patients with a CVD event within the past six months. Although the results from FAST were reassuring, the use of febuxostat in patients with prior CVD should be considered on an individual basis.
Probenecid, a uricosuric medication, is prescribed for patients who are intolerant or nonresponsive to allopurinol and can be used as monotherapy or in combination with a xanthine oxidase inhibitor. However, probenecid is contraindicated in people with a history of nephrolithiasis and it has reduced efficacy in those with renal impairment.32 In patients who are nonresponsive to xanthine oxidase inhibitors, referral to a rheumatologist is recommended for consideration of uricosuric medications.
Flare prophylaxis and urate-lowering therapy
Patients with gout should be prescribed concomitant flare prophylaxis at ULT initiation and until the target serum urate level is achieved.32,33 This is usually for a duration of three to six months, but may be longer in patients with frequent flares or advanced gout. Colchicine is the preferred prophylactic as it has a better safety profile compared with NSAIDs or corticosteroids. However, NSAIDs or corticosteroids (e.g. naproxen 500 mg daily or prednisone 5 mg daily) may be appropriate for patients who are intolerant to colchicine, but careful monitoring is required with consideration of relevant patient comorbidities. Every patient with gout should have an action plan for future gout flares that allows them to initiate treatment as soon as possible.
Adjuncts to urate-lowering therapy and new therapies
Medications, including fenofibrate, losartan and sodium-glucose cotransporter-2 inhibitors, have been shown to have a modest urate lowering effect.39-41 New anti-inflammatory therapies include the interleukin-1 beta inhibitors anakinra and canakinumab. Anakinra is approved by the TGA and is indicated for use in moderate to severe cryopyrin associated periodic syndromes (CAPS); canakinumab was recently approved by the US Food and Drug Administration but is not approved by the TGA. Both are used off-label for severe gout, although their use is limited by cost and availability. Pegloticase, a pegylated recombinant uricase administered as a fortnightly intravenous infusion, shows promise as an option for patients with refractory gout; however, it is not approved for use in Australia.42
Future directions for gout management
Despite highly effective therapy, gout remains undertreated. An Australian study found only 22% of patients achieved a serum urate concentration below 0.36 mmol/L over a five year period.4 Nurse- and pharmacist-led gout management programs with protocolised ULT have been shown to be more effective than traditional doctor-led models of care and highlight the importance of education and the provision of time to allow exploration of patient concerns and barriers to treatment.43,44 Telehealth appointments and gout-specific phone apps may also improve self-management.45
Calcium pyrophosphate deposition disease
An overview of CPPD
CPPD has an estimated prevalence of between 5 and 7% according to radiographic studies from the UK and US.46,47 However, the prevalence of CPPD in the Australian population is unknown.
The term ‘pseudogout’ was adopted following initial descriptions of CPP crystals found in synovial fluid of people with episodes of acute monoarthritis typical of gout but with normal serum urate levels.48 However, further studies demonstrated the presence of CPP crystals in other musculoskeletal conditions, with the subsequent description of ‘pseudo-osteoarthritis’ and ‘pseudo-rheumatoid arthritis’ in the literature.49 To reduce confusion and improve consistency of descriptive terms for this disease, CPPD is now used as an umbrella term which includes acute CPP crystal arthritis (formerly known as pseudogout), chronic CPP crystal inflammatory arthritis, osteoarthritis with CPPD and asymptomatic chondrocalcinosis.50 There can be considerable overlap across these phenotypes.
Clinical presentations of CPPD
Acute CPP crystal arthritis
The most commonly recognised presentation of CPPD is acute CPP crystal arthritis, which typically presents as a monoarticular or oligoarticular arthritis. This usually manifests with pain, stiffness, erythema and swelling developing over a few hours, and can be indistinguishable from gout or septic arthritis. Systemic symptoms, including headache, neck pain and fever (estimated to affect 50% of people with CPPD), can also occur.51
In contrast to acute flares of gout, which typically resolve within several days to one week, acute CPP crystal arthritis may persist for weeks, is usually less painful and slower to reach peak intensity. Most episodes are self-limiting and resolve within one to three weeks. The most commonly involved joint is the knee, followed by the wrist but may also involve the ankle, elbow, toe, shoulder and hip. Rarely, CPP crystals can deposit in the spine, symphysis pubis, the temporomandibular joint, tendons and bursae.52 CPPD in the cervical spine can also cause crowned dens syndrome, a rare cause of acute neck pain.53 Most episodes of acute CPP crystal arthritis develop spontaneously but can be precipitated by trauma or intercurrent illness.
Osteoarthritis with CPPD and asymptomatic chondrocalcinosis
CPPD is often seen in the context of osteoarthritis as the prevalence of both conditions increases with age. However, relative to patients with osteoarthritis alone, those with osteoarthritis and CPPD tend to have more intense pain, joint tenderness and systemic symptoms, including fever. Additionally, osteoarthritis with CPPD has a different distribution of affected joints, including isolated involvement of the wrist, elbow, shoulder or ankle, which is not seen in osteoarthritis alone.54 Asymptomatic chondrocalcinosis occurs in patients with radiographic evidence of CPPD on incident x-rays but with no apparent clinical symptoms.
Chronic CPP crystal inflammatory arthritis
This form of CPPD has several clinical phenotypes, with polyarticular involvement resembling osteoarthritis and, rarely, rheumatoid arthritis. The osteoarthritis phenotype is characterised by flares of inflammatory symptoms and signs but can affect joints less commonly involved in typical osteoarthritis, including the glenohumeral, wrist and metacarpophalangeal joints.55 The rheumatoid arthritis phenotype usually involves large and small joints; however, unlike typical rheumatoid arthritis, is usually less symmetrical. Chronic CPP crystal inflammatory arthritis is more frequently seen in younger patients (age <50 years) with a familial predisposition to CPPD.55
Risk factors for CPPD
Risk factors for CPPD include increasing age, previous joint injury or surgery, a history of gout, a family history of CPPD and endocrine and metabolic conditions. The risk of CPPD increases with age, and beyond the age of 60 years, the likelihood of developing CPPD doubles with every decade.56 CPPD may be more common in women, with a recent study finding women were twice as likely to have acute CPP crystal arthritis.57 People under 50 years of age diagnosed with CPPD should be screened for conditions including haemochromatosis, hyperparathyroidism, hypomagnesaemia and hypophosphatasia, and a detailed family history taken to establish a potential familial predisposition to this disease.58-60 There are no known dietary associations with CPPD.55
Pathophysiology of CPPD
The pathways by which deposition of CPP crystals cause joint inflammation and damage are not fully understood. Chondrocytes produce high levels of extracellular inorganic pyrophosphate, which is crucial to the formation of CPP crystals and may explain why CPP crystals are rarely found in noncartilaginous tissues.61 In cartilaginous tissues, ANKH membrane proteins regulate inorganic pyrophosphate production from extracellular adenosine triphosphate.62 Mutations in the ANKH membrane protein gene have been implicated in familial CPPD.52 Inorganic pyrophosphate binds to calcium to form CPP crystals in the cartilage and synovium of joints. CPP crystals activate the NLRP3 inflammasome (similar to gout) and generate destructive matrix metalloproteinases and altered cartilage mechanics, which initiates inflammation and joint damage.63,64
Diagnosis of CPPD
CPPD can be difficult to diagnose on history and clinical examination alone because it presents similarly to gout, septic arthritis and other forms of inflammatory arthritis. It is important to establish the number and site of the affected joints, the characteristics of the symptoms, any precipitating factors, relevant comorbidities and family history. When possible, a joint aspirate should be obtained and sent for microscopy, culture and crystal analysis. This is particularly important in an acute monoarthritis to exclude septic arthritis, although CPPD can occur concurrently with infection. The gold standard for definitive diagnosis of CPPD is the identification of positively birefringent, rhomboid or rod-shaped crystals on polarised light microscopy.55 MSU crystals may also be present, and the patient may require treatment for both gout and CPPD.
Blood tests
Patients with CPPD may show leukocytosis on a full blood count and their C-reactive protein levels may be elevated. Renal function should be assessed to determine whether NSAIDs are contraindicated. Serum urate levels can be elevated in 20% of patients with CPPD and, although a normal result may reduce the likelihood of gout, testing should be repeated when the patient is asymptomatic. In younger patients (aged <55 years) with suspected CPPD, parathyroid hormone, calcium, phosphate, magnesium and iron levels should be assessed to screen for metabolic conditions.
Imaging
In cases where obtaining synovial fluid is challenging, plain radiography, ultrasonography and DECT may be helpful to identify CPPD. The characteristic finding on radiography is chondrocalcinosis, which appears as punctate or linear opacities in the fibrocartilage or hyaline articular cartilage regions. (Figure 2).65 If radiology is performed, x-rays should include the wrists and the pelvis (anteroposterior view) in addition to the knees, as x-rays of knees alone may miss CPPD. The absence of chondrocalcinosis on x-ray does not exclude CPPD.50

Ultrasound and CT imaging can be useful in patients with atypical symptoms or rare presentations. Ultrasound has been shown to be more sensitive but less specific for identifying CPP crystals and can also be used to guide joint aspiration.66 CT can be used to diagnose CPPD deposition in the spine and neck (crowned den syndrome). DECT is able to distinguish between CPP and MSU crystals but may not be readily accessible.67
Management of CPPD
In contrast to gout, there are currently no effective disease-modifying therapies that reduce or remove CPP crystals in joints. Furthermore, treatment is not indicated for patients who have asymptomatic chondrocalcinosis. In patients with CPPD secondary to an underlying metabolic condition (e.g. haemochromatosis, primary hyperparathyroidism), treatment should focus on correcting the underlying condition. The management of CPPD can be divided into treatment of acute CPP crystal arthritis and chronic CPP crystal inflammatory arthritis. In patients with osteoarthritis with CPPD, management should follow clinical guidelines for osteoarthritis.68
Acute CPP crystal arthritis
Nonpharmacological management includes use of ice packs and taking rest. Joint aspiration may temporarily reduce pain due to reduction in the fluid volume within the joint. Pharmacological management can be localised or systemic. Localised therapy in the form of an intra-articular corticosteroid injection can be effective in reducing symptoms in those with a single joint affected.
Systemic therapy includes oral NSAIDs, colchicine or oral corticosteroids, the choice of which will take into consideration patient age, renal function, comorbidities and the risk of adverse effects (Table 2). Recently, the first randomised controlled trial comparing colchicine with oral prednisone for the management of acute CPP showed that both had equivalent short-term efficacy and safety profiles.69 The duration of treatment of systemic therapies usually depends on symptom relief and adverse effects. Concomitant proton pump inhibitor therapy should be considered for patients prescribed NSAIDs for gastric protection. Where it is deemed appropriate and safe, low dose colchicine may be effective in reducing the frequency of episodes in patients with recurrent acute CPP crystal arthritis.70

Chronic CPP crystal inflammatory arthritis
Because of the lack of disease-modifying therapies and limited clinical trials on targeted treatments for chronic CPP crystal inflammatory arthritis, management is primarily directed towards reducing inflammation. Individual patient factors need to be considered before the long-term prescription of NSAIDs, low dose colchicine (0.5 to 1 mg daily) or low dose oral corticosteroids. Long-term use of these medications requires ongoing patient monitoring for adverse effects and drug toxicity. Patients taking regular colchicine should undergo routine monitoring of renal function, a full blood count (for leucopenia and thrombocytopenia) and creatine kinase levels (colchicine-induced myotoxicity). There is some evidence for the use of the immunomodulator methotrexate in patients with chronic CPP crystal inflammatory arthritis that is refractory to NSAIDs; however, a rheumatologist review is recommended before considering these options.71
Progressive joint damage can result from both chronic CPP crystal inflammatory arthritis and recurrent acute CPP crystal arthritis, and can occur in combination with osteoarthritis. Therefore, where appropriate, patients should be considered for joint replacement surgery, taking the same approach as for patients with osteoarthritis alone. Similar to gout, two recent studies have shown a significant association between CPPD and CVD, with research on this relationship ongoing.72,73
Future directions for CPPD research
To facilitate future research across this heterogeneous disease, the American College of Rheumatology and European Alliance of Associations for Rheumatology have developed the first validated classification criteria for symptomatic CPPD.74 In addition, the Outcome Measures in Rheumatology (OMERACT) group have developed CPPD Core Domain Sets to help define the important outcomes to be measured in future clinical trials.75 There is considerable interest in therapies that may lead to CPP crystal dissolution, with histone deacetylase inhibitors recently shown to downregulate CPP crystal formation in human articular chondrocytes.76
Conclusion
Gout and CPPD are the most common forms of crystal arthritis encountered in primary care. Confirmation of MSU or CPP crystals in synovial fluid using polarising light microscopy remains the gold standard for diagnosis but clinical history and imaging may be of assistance. Management of gout includes treatment of the acute flare and ULT. Anti-inflammatory therapies are the mainstay of treatment for CPPD as, currently, there are no disease modifying therapies available. MT
COMPETING INTERESTS: None.
References
1. Ofori-Asenso R, Ilomaki J, Curtis AJ, Zomer E, Zoungas S, Liew D. Patterns of medication dispensation for multiple comorbidities among older adults in Australia. Pharmacy (Basel) 2018; 6: 134.
2. Pathmanathan K, Robinson PC, Hill CL, Keen HI. The prevalence of gout and hyperuricaemia in Australia: an updated systematic review. Semin Arthritis Rheum 2021; 51: 121-128.
3. Proudman C, Lester SE, Gonzalez-Chica DA, Gill TK, Dalbeth N, Hill CL. Gout, flares, and allopurinol use: a population-based study. Arthritis Res Ther 2019; 21: 132.
4. Robinson PC, Taylor WJ, Dalbeth N. An observational study of gout prevalence and quality of care in a national Australian general practice population. J Rheumatol 2015; 42: 1702-1707.
5. Nguyen AD, Lind KE, Day RO, Georgiou A, Westbrook JI. A profile of health status and demographics of aged care facility residents with gout. Australas J Ageing 2020; 39: e153-e161.
6. Winnard D, Wright C, Taylor WJ, et al. National prevalence of gout derived from administrative health data in Aotearoa New Zealand. Rheumatology (Oxford) 2012; 51: 901-909.
7. Wong PKK, Ng BCK, Mitchell J, et al. The disproportionately large contribution of the Māori and Pacific Islander community to the healthcare burden of gout in Western Sydney. Intern Med J 2023; 53: 1450-1457.
8. Coleman W, Spencer D, Wong P, Manolios N. An enquiry into the crippling gout affecting Pacific Islander and Māori men in Western Sydney. Int J Rheum Dis 2021; 24: 1394-1401.
9. Coulshed A, Nguyen AD, Stocker SL, Day RO. Australian patient perspectives on the impact of gout. Int J Rheum Dis 2020; 23: 1372-1378.
10. Australian Institute of Health and Welfare (AIHW). Chronic musculoskeletal conditions: Gout. AIHW; Canberra, 2023. Available online at https://www.aihw.gov.au/reports/chronic-musculoskeletal-conditions/gout (accessed March 2024).
11. Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007-2008. Am J Med 2012; 125(7): 679-687.e1.
12. Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 1987; 82: 421-426.
13. Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet 2021; 15: 1843-1855.
14. Major TJ, Topless RK, Dalbeth N, Merriman TR. Evaluation of the diet wide contribution to serum urate levels: meta-analysis of population-based cohorts. BMJ 2018; 363: k3951.
15. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med 2004; 350: 1093-1103.
16. Choi HK, Atkinson K, Karlson EW, Willett W, Curhan G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet 2004; 363: 1277-1281.
17. Choi HK, Curhan G. Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ 2008; 336: 309-312.
18. Tanner C, Boocock J, Stahl EA, et al. Population-specific resequencing associates the ATP-binding cassette subfamily C member 4 gene with gout in New Zealand Māori and Pacific Men. Arthritis Rheumatol 2017; 69: 1461-1469.
19. Angell B, Laba TL, Lung T, et al. Healthcare expenditure on Indigenous and non-Indigenous Australians at high risk of cardiovascular disease. Int J Equity Health 2017; 16: 108.
20. Lee G, Roberts L. Healthcare burden of in-hospital gout. Intern Med J 2012; 42: 1261-1263.
21. Minaur N, Sawyers S, Parker J, Darmawan J. Rheumatic disease in an Australian Aboriginal community in North Queensland, Australia. A WHO-ILAR COPCORD survey. J Rheumatol 2004; 31: 965-972.
22. Dalbeth N, Phipps-Green A, Frampton C, Neogi T, Taylor WJ, Merriman TR. Relationship between serum urate concentration and clinically evident incident gout: an individual participant data analysis. Ann Rheum Dis 2018; 77: 1048-1052.
23. Chhana A, Lee G, Dalbeth N. Factors influencing the crystallization of monosodium urate: a systematic literature review. BMC Musculoskelet Disord 2015; 16: 296.
24. Palmer DG, Highton J, Hessian PA. Development of the gout tophus. An hypothesis. Am J Clin Pathol 1989; 91: 190-195.
25. Janssens HJ, Fransen J, van de Lisdonk EH, van Riel PL, van Weel C, Janssen M. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med 2010; 170: 1120-1126.
26. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol 2002; 29: 1950-1953.
27. Lee YH, Song GG. Diagnostic accuracy of dual-energy computed tomography in patients with gout: a meta-analysis. Semin Arthritis Rheum 2017; 47: 95-101.
28. Lee YH, Song GG. Diagnostic accuracy of ultrasound in patients with gout: a meta-analysis. Semin Arthritis Rheum 2018; 47: 703-709.
29. Christiansen SN, Filippou G, Scire CA, et al. Consensus-based semi-quantitative ultrasound scoring system for gout lesions: Results of an OMERACT Delphi process and web-reliability exercise. Semin Arthritis Rheum 2021; 51: 644-649.
30. Dessein PH, Shipton EA, Stanwix AE, Joffe BI, Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Ann Rheum Dis 2000; 59: 539-543.
31. Schlesinger N, Detry MA, Holland BK, et al. Local ice therapy during bouts of acute gouty arthritis. J Rheumatol 2002; 29: 331-334.
32. Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis 2017; 76: 29-42.
33. FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Rheumatol 2020; 72: 879-895.
34. Hill EM, Sky K, Sit M, Collamer A, Higgs J. Does starting allopurinol prolong acute treated gout? A randomized clinical trial. J Clin Rheumatol 2015; 21: 120-125.
35. Taylor TH, Mecchella JN, Larson RJ, Kerin KD, Mackenzie TA. Initiation of allopurinol at first medical contact for acute attacks of gout: a randomized clinical trial. Am J Med 2012; 125: 1126-1134.e7.
36. Keller SF, Lu N, Blumenthal KG, et al. Racial/ethnic variation and risk factors for allopurinol-associated severe cutaneous adverse reactions: a cohort study. Ann Rheum Dis 2018; 77: 1187-1193.
37. Mackenzie IS, Ford I, Nuki G, et al. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (FAST): a multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet 2020; 396: 1745-1757.
38. White WB, Saag KG, Becker MA, et al. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N Engl J Med 2018; 378: 1200-1210.
39. Choi HK, Soriano LC, Zhang Y, Rodriguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344: d8190.
40. Waldman B, Ansquer JC, Sullivan DR, et al. Effect of fenofibrate on uric acid and gout in type 2 diabetes: a post-hoc analysis of the randomised, controlled FIELD study. Lancet Diabetes Endocrinol 2018; 6: 310-318.
41. Li J, Badve SV, Zhou Z, et al. The effects of canagliflozin on gout in type 2 diabetes: a post-hoc analysis of the CANVAS Program. Lancet Rheumatol 2019; 1: e220-e228.
42. Sundy JS, Baraf HS, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA 2011; 306: 711-720.
43. Doherty M, Jenkins W, Richardson H, et al. Efficacy and cost-effectiveness of nurse-led care involving education and engagement of patients and a treat-to-target urate-lowering strategy versus usual care for gout: a randomised controlled trial. Lancet 2018; 392: 1403-1412.
44. Huang IJ, Liew JW, Morcos MB, Zuo S, Crawford C, Bays AM. Pharmacist-managed titration of urate-lowering therapy to streamline gout management. Rheumatol Int 2019; 39: 1637-1641.
45. Day RO, Frensham LJ, Nguyen AD, et al. Effectiveness of an electronic patient-centred self-management tool for gout sufferers: a cluster randomised controlled trial protocol. BMJ Open 2017; 7: e017281.
46. Felson DT, Naimark A, Anderson J, Kazis L, Castelli W, Meenan RF. The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum 1987; 30: 914-978.
47. Neame RL, Carr AJ, Muir K, Doherty M. UK community prevalence of knee chondrocalcinosis: evidence that correlation with osteoarthritis is through a shared association with osteophyte. Ann Rheum Dis 2003; 62: 513-518.
48. McCarty D, Kohn NN, Faires JS. The significance of calcium pyrophosphate crystals in the synovial fluid of arthritic patients: The "pseudogout syndrome": 1. Clinical Aspects. Ann Intern Med 1962; 56: 711-737.
49. Zhang W, Doherty M, Peat G, et al. EULAR evidence-based recommendations for the diagnosis of knee osteoarthritis. Ann Rheum Dis 2010; 69: 483-489.
50. Zhang W, Doherty M, Bardin T, et al. European League Against Rheumatism recommendations for calcium pyrophosphate deposition. Part I: terminology and diagnosis. Ann Rheum Dis 2011; 70: 563-570.
51. Masuda I, Ishikawa K. Clinical features of pseudogout attack. A survey of 50 cases. Clin Orthop Relat Res 1988; (229): 173-181.
52. Abhishek A, Doherty M. Pathophysiology of articular chondrocalcinosis--role of ANKH. Nat Rev Rheumatol 2011; 7: 96-104.
53. Bouvet JP, le Parc JM, Michalski B, Benlahrache C, Auquier L. Acute neck pain due to calcifications surrounding the odontoid process: the crowned dens syndrome. Arthritis Rheum 1985; 28 1417-1420.
54. Rosenthal AK, Ryan LM. Calcium pyrophosphate deposition disease. N Engl J Med 2016; 374: 2575-2584.
55. Zitnan D, Sit’Aj S. Chondrocalcinosis articularis. Section 1. Clinical and radiological study. Ann Rheum Dis 1963; 22: 142-152.
56. Wilkins E, Dieppe P, Maddison P, Evison G. Osteoarthritis and articular chondrocalcinosis in the elderly. Ann Rheum Dis 1983; 42: 280-284.
57. Yates KA, Yoshida K, Xu C, et al. Acute calcium pyrophosphate crystal arthritis flare rate and risk factors for recurrence. J Rheumatol 2020; 47: 1261-1266.
58. Jones AC, Chuck AJ, Arie EA, Green DJ, Doherty M. Diseases associated with calcium pyrophosphate deposition disease. Semin Arthritis Rheum 1992; 22: 188-202.
59. Pritchard MH, Jessop JD. Chondrocalcinosis in primary hyperparathyroidism. Influence of age, metabolic bone disease, and parathyroidectomy. Ann Rheum Dis 1977; 36: 146-151.
60. Richette P, Ayoub G, Lahalle S, et al. Hypomagnesemia associated with chondrocalcinosis: a cross-sectional study. Arthritis Rheum 2007; 57: 1496-1501.
61. Ryan LM, Rosenthal AK. Metabolism of extracellular pyrophosphate. Curr Opin Rheumatol 2003; 15: 311-314.
62. Rosenthal AK, Gohr CM, Mitton-Fitzgerald E, Lutz MK, Dubyak GR, Ryan LM. The progressive ankylosis gene product ANK regulates extracellular ATP levels in primary articular chondrocytes. Arthritis Res Ther 2013; 15: R154.
63. Muehleman C, Li J, Aigner T, et al. Association between crystals and cartilage degeneration in the ankle. J Rheumatol 2008; 35: 1108-1117.
64. Reuben PM, Wenger L, Cruz M, Cheung HS. Induction of matrix metalloproteinase-8 in human fibroblasts by basic calcium phosphate and calcium pyrophosphate dihydrate crystals: effect of phosphocitrate. Connect Tissue Res 2001; 42: 1-12.
65. Tedeschi SK, Becce F, Pascart T, et al. Imaging features of calcium pyrophosphate deposition disease: consensus definitions from an international multidisciplinary working group. Arthritis Care Res (Hoboken) 2023; 75: 825-834.
66. Cipolletta E, Filippou G, Scire CA, et al. The diagnostic value of conventional radiography and musculoskeletal ultrasonography in calcium pyrophosphate deposition disease: a systematic literature review and meta-analysis. Osteoarthritis Cartilage 2021; 29: 619-632.
67. Filippou G, Filippucci E, Mandl P, Abhishek A. A critical review of the available evidence on the diagnosis and clinical features of CPPD: do we really need imaging? Clin Rheumatol 2021; 40: 2581-2592.
68. Royal Australian College of General Practitioners (RACGP). Guideline for the management of knee and hip osteoarthritis. Second ed. East Melbourne, Vic: RACGP, 2018. Available online at: https://www.racgp.org.au/clinical-resources/clinical-guidelines/key-racgp-guidelines/view-all-racgp-guidelines/knee-and-hip-osteoarthritis (accessed March 2024).
69. Pascart T, Robinet P, Ottaviani S, et al. Evaluating the safety and short-term equivalence of colchicine versus prednisone in older patients with acute calcium pyrophosphate crystal arthritis (COLCHICORT): an open-label, multicentre, randomised trial. Lancet Rheumatol 2023; 5: e523-e531.
70. Zhang W, Doherty M, Pascual E, et al. EULAR recommendations for calcium pyrophosphate deposition. Part II: management. Ann Rheum Dis 2011; 70: 571-575.
71. Chollet-Janin A, Finckh A, Dudler J, Guerne PA. Methotrexate as an alternative therapy for chronic calcium pyrophosphate deposition disease: an exploratory analysis. Arthritis Rheum 2007; 56: 688-692.
72. Bashir M, Sherman KA, Solomon DH, Rosenthal A, Tedeschi SK. Cardiovascular disease risk in calcium pyrophosphate deposition disease: a nationwide study of veterans. Arthritis Care Res (Hoboken) 2023; 75: 277-282.
73. Tedeschi SK, Huang W, Yoshida K, Solomon DH. Risk of cardiovascular events in patients having had acute calcium pyrophosphate crystal arthritis. Ann Rheum Dis 2022; annrheumdis-2022-222387.
74. Abhishek A, Tedeschi SK, Pascart T, et al. The 2023 ACR/EULAR classification criteria for calcium pyrophosphate deposition disease. Ann Rheum Dis 2023; 82: 1248-1257.
75. Cai K, Fuller A, Zhang Y, et al. Towards development of core domain sets for short term and long term studies of calcium pyrophosphate crystal deposition (CPPD) disease : a framework paper by the OMERACT CPPD Working Group. Semin Arthritis Rheum 2021; 51: 946-950.
76. Chang CC, Lee KL, Chan TS, Chung CC, Liang YC. Histone deacetylase inhibitors downregulate calcium pyrophosphate crystal formation in human articular chondrocytes. Int J Mol Sci 2022; 23: 2604.
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.